Unusual Sites of Leiomyosarcoma: A Study of 3 Interesting Cases
By Bushra Siddiqui1, Shahbaz Habib Faridi2, Asfa Shams1, Jowairiah Hassan1Affiliations
doi: 10.29271/jcpsp.2022.09.1205ABSTRACT
Leiomyosarcoma malignant tumours arise from the smooth muscle cells. They are rapidly growing tumours with aggressive behaviour and poor prognosis. Although relatively rare, they pose a diagnostic challenge as they comprise a large spectrum of diagnostic entities. Herein, we describe three cases of leiomyosarcoma developing at unusual sites and posing diagnostic challenges. Our first case is a leiomyosarcoma developing at the post-burn scar site; the second case is of primary pulmonary leiomyosarcoma, and the third is retroperitoneal leiomyosarcoma. Histopathological examination is not sufficient all the time for making the diagnosis of leiomyosarcoma because there is a morphologic overlap with other malignancies. Immunohistochemistry acts as an adjunct to arrive at a definite diagnosis and hence, proves to be the most important ancillary technique in the diagnosis of such tumours. Though these tumours arise most commonly from the uterine smooth muscle, but rarely encountered at unusual sites posing diagnostic difficulties.
Key Words: Leiomyosarcoma, Retroperitoneal, Pulmonary, Post-burn scar.
INTRODUCTION
Leiomyosarcomas are malignant tumours arising from smooth muscle cells. These are rare tumours comprising 5% to 10% of all soft tissue sarcomas.1 Soft tissue leiomyosarcomas are less common than uterine and gastric leiomyosarcomas.2 Since the site is an important factor in assessing outcome, soft tissue leiomyosarcomas are divided into different subgroups depending on their site. The most common amongst these are leiomyosarcomas of retroperitoneum and abdominal cavity; followed by leiomyosarcomas of somatic soft tissue, cutaneous leiomyosarcomas, and leiomyosarcomas of vascular origin.2 Sarcomas arising in unusual locations i.e. pulmonary leiomyosarcomas and sarcomas arising in the post-burn scar have rarely been described in the literature.3,4 This article reports a series of 3 cases of leiomyosarcoma presenting at the unusual sites and posing a diagnostic difficulty.
CASE 1:
A 55-year female presented with post-burn ulceration of the thigh. The patient, who was otherwise apparently healthy, had a history of burn over anterior aspect of the left thigh 8 years ago.
The burn had healed but for the last five months, the patient had noticed an ulcerated growth developing over the scar. A provisional diagnosis of dermatofibrosarcoma protuberans was made. Excision of the growth was done and a partially covered skin tissue piece with a nodular growth over its skin surface was received. The whole specimen measured 18 ×16×4 cm with ellipse of skin measuring 17×4 cm and the nodular growth measuring 6×6 cm. The growth was 2 cm above the skin surface. On sectioning, a well-circumscribed creamish white, firm area was seen measuring 7.5×6.5×3.6 cm. The tumour was extending into the deeper tissue and reaching up to the base.
Microscopically, sections showed stratified squamous epithelium with a well-circumscribed lesion in the superficial dermis extending upto the deep reticular dermis with no extension into the subcutaneous fat. The tumour cells were arranged in broad intersecting fascicles and showed mild to moderate pleomorphism having oval to cigar-shaped nuclei with vesicular chromatin, prominent nucleoli, and eosinophilic fibrillary cytoplasm (Figure 1a and b). Stroma showed myxoid change and proliferation of the blood vessels. The mitotic count was 8-9/10 HPF. No necrosis was seen. Margins were free from the tumour while the base was involved by the tumour cells. Immunohistochemistry (IHC), for vimentin and smooth muscle actin (SMA), was positive (Figure 1c). CD 34 was negative. A diagnosis of cutaneous leiomyosarcoma, French Federation of Cancer Centres Sarcoma Group (FNCLCC) grade1, was made.
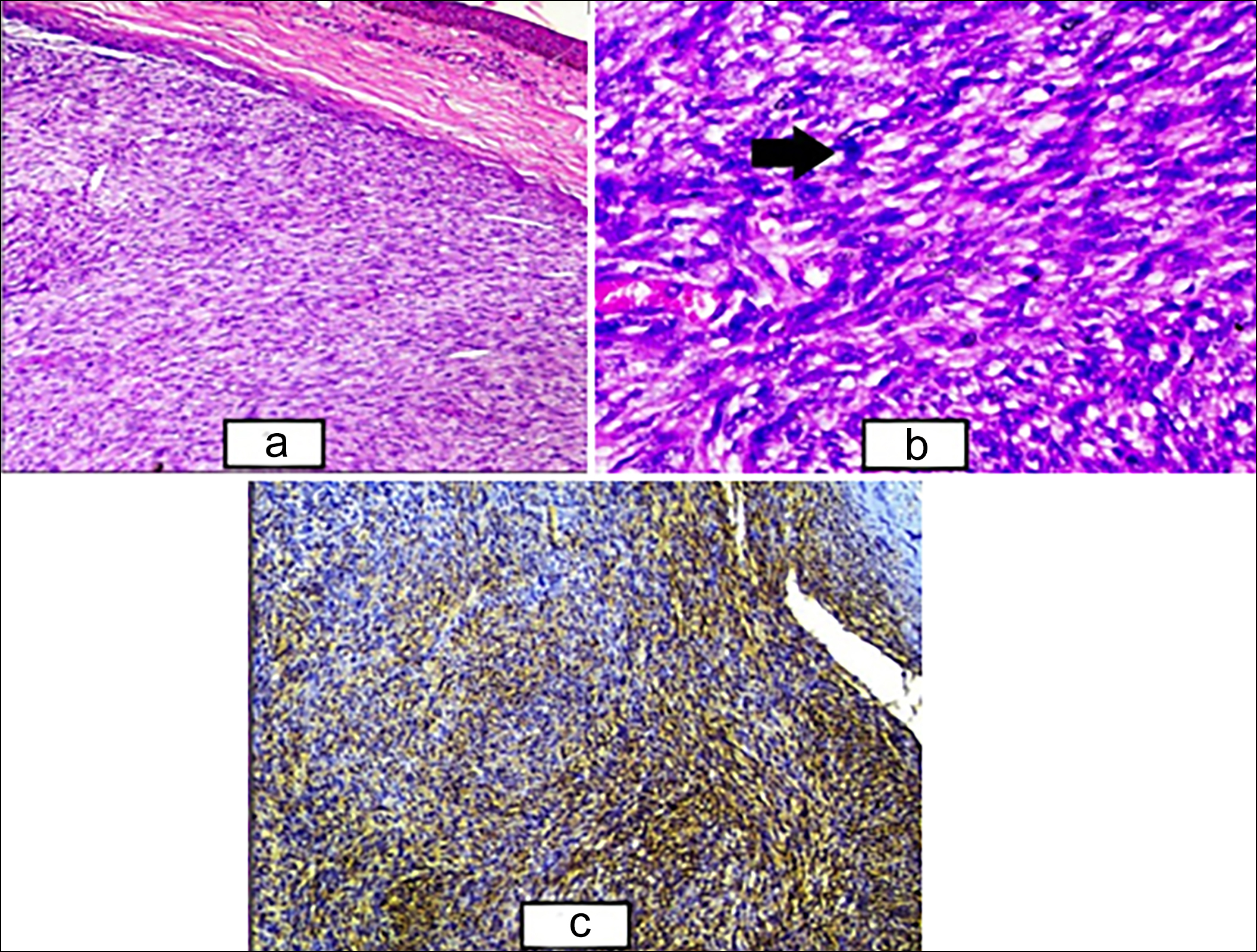 Figure 1: (a) Epidermal covering seen along with dermis showing the sheets and fascicles of atypical smooth muscle fibres (H&E, ×100). (b) Higher power showing the sheets and fascicles of smooth muscle fibres with nuclear atypia (black arrow) and hyperchromasia along with congested blood vessel (H&E, ×400). (c) Cytoplasmic positivity of SMA seen in tumour cells (SMA, ×100).
Figure 1: (a) Epidermal covering seen along with dermis showing the sheets and fascicles of atypical smooth muscle fibres (H&E, ×100). (b) Higher power showing the sheets and fascicles of smooth muscle fibres with nuclear atypia (black arrow) and hyperchromasia along with congested blood vessel (H&E, ×400). (c) Cytoplasmic positivity of SMA seen in tumour cells (SMA, ×100).
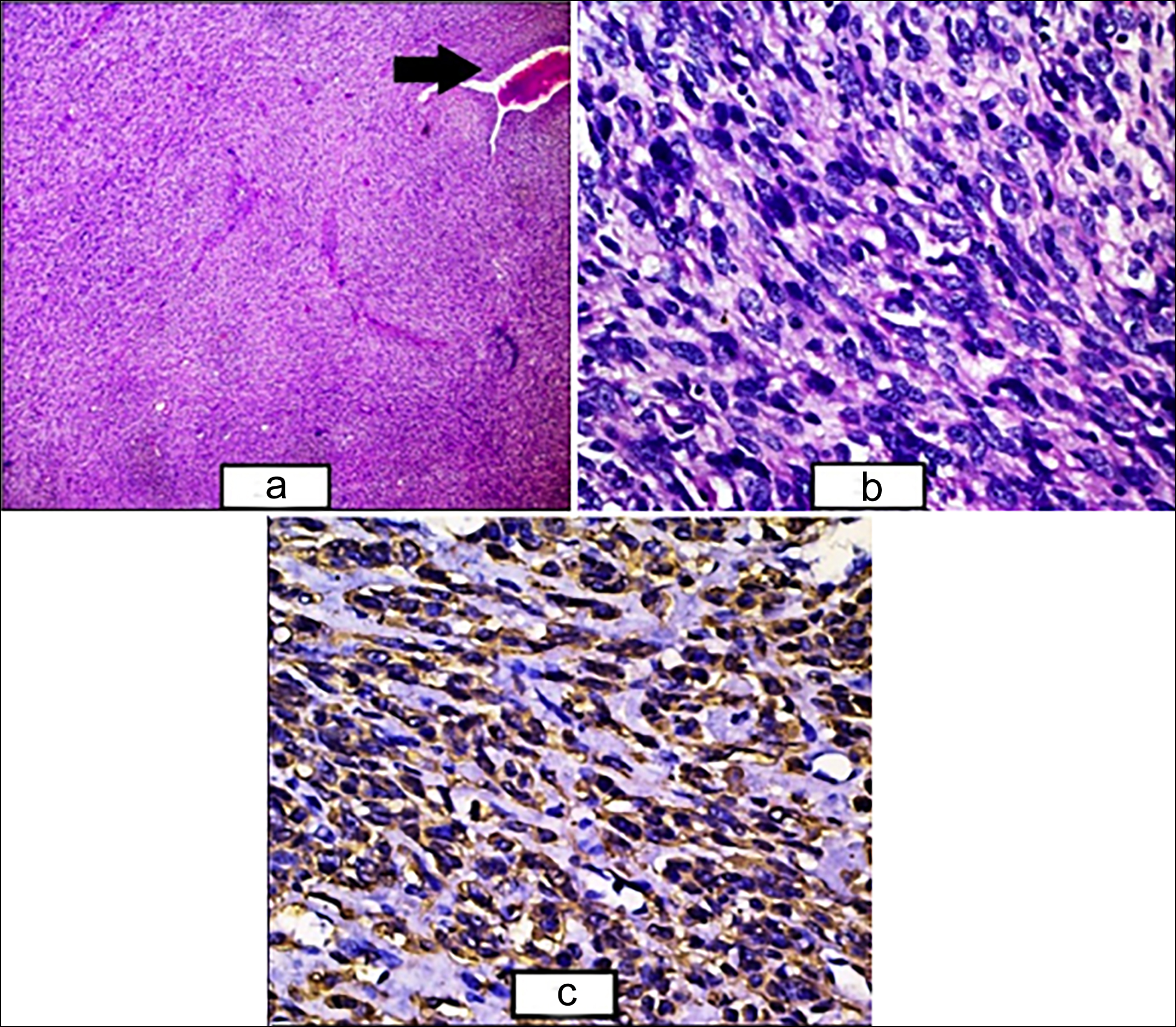 Figure 2: (a) The sheets and fascicles of atypical smooth muscle fibres along with congested hemagiopericytomatous blood vessel (black arrow) (H&E, ×40). (b) Higher power showing the sheets and fascicles of smooth muscle fibres with nuclear atypia and hyperchromasia along with few bizarre nuclei and apoptotic bodies (H&E, ×400). (c) Cytoplasmic positivity of SMA seen in tumour cells (SMA, ×400).
Figure 2: (a) The sheets and fascicles of atypical smooth muscle fibres along with congested hemagiopericytomatous blood vessel (black arrow) (H&E, ×40). (b) Higher power showing the sheets and fascicles of smooth muscle fibres with nuclear atypia and hyperchromasia along with few bizarre nuclei and apoptotic bodies (H&E, ×400). (c) Cytoplasmic positivity of SMA seen in tumour cells (SMA, ×400).
CASE 2:
The second case was of a 30-year male who presented with the right-sided chest pain and hemoptysis of 6 months duration. Computed Tomography (CT) findings showed a posterior mediastinal mass with lytic destruction of 9th and 10th ribs and on FDG scan, a large hypodense soft tissue density mass lesion measuring13×11×8.2 cm with areas of necrosis was seen in the right lung lower lobe. The mass was abutting the right lower bronchus, costal, and diaphragmatic pleura on the right side and also eroding the right 9th and 10th ribs posteriorly. The biopsy showed a predominantly diffuse infiltrate of round-to-oval cells with ill-defined cytoplasm and focal vague acinar pattern in a vascular stroma. A differential diagnosis of lymphoma, small cell variant of squamous cell carcinoma (SCC), and peripheral neuroectodermal tumour (PNET)/ Ewing sarcoma was made. However, IHC staining for CD45, chromogranin, B and T cell markers were negative. Biopsy was repeated, and a diagnosis of small cell carcinoma was made and the patient underwent chemotherapy for the same. Repeat imaging showed necrotic foci in the tumour with slight reduction in the size. A right hemilobectomy was performed, and the specimen was sent for histopathology. Grossly, black-coloured partly necrotic lung tissue measuring 7×6×5 cm was received which on sectioning showed mucoid areas admixed with necrotic areas. No mediastinal impression was identified. Two ribs were received with attached soft tissue. Representative sections were taken. On hematoxylin and eosin staining, sections showed hyper cellular as well as hypocellular myxoid areas along with areas of whirling. The cells were round-to-oval with vesicular nuclei and prominent eosinophilic nucleoli and eosinophilic cytoplasm. Multi-nucleation was also seen along with the areas of vascular proliferation (Figures 2 a and b). Mitotic count was > 10/10 HPF, and necrosis was <50% of the total area. Soft tissue that was scraped off from the ribs was involved by the tumour; however, ribs were uninvolved. IHC staining for vimentin and SMA showed cytoplasmic positivity (Figure 2c) while epithelial membrane antigen (EMA), desmin, and pan-CK were negative. A final diagnosis of primary pulmonary leiomyosarcoma, FNCLCC grade 2, was made.
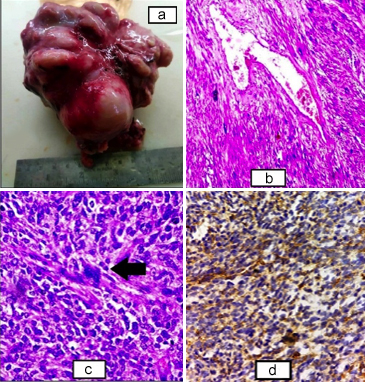 Figure 3: (a) Gross specimen of retroperitoneal mass, solid, and nodular outer surface with the areas of congestion. (b) Sheets and fascicles of the atypical smooth muscle fibres along with congested hemagiopericytomatous blood vessel (H&E, ×100). (c) Higher power showing the sheets and fascicles of smooth muscle fibres with nuclear atypia and hyperchromasia along with tumour giant cells (black arrow) (H&E, ×400). (d) Cytoplasmic positivity of SMA seen in the tumour cells (SMA, ×400).
Figure 3: (a) Gross specimen of retroperitoneal mass, solid, and nodular outer surface with the areas of congestion. (b) Sheets and fascicles of the atypical smooth muscle fibres along with congested hemagiopericytomatous blood vessel (H&E, ×100). (c) Higher power showing the sheets and fascicles of smooth muscle fibres with nuclear atypia and hyperchromasia along with tumour giant cells (black arrow) (H&E, ×400). (d) Cytoplasmic positivity of SMA seen in the tumour cells (SMA, ×400).
CASE 3:
A 36-year female was admitted with dull aching pain in abdomen for 1 year and abdominal lump for 6 months. There were no menstrual complaints. On ultrasonography, a retroperitoneal mass with numerous sub-serosal fibroids was present. A presumptive diagnosis of lymphoma was made and the mass was resected. Imprint smears were also sent. On cytopathology, a diagnosis of malignant spindle cell neoplasm was made. Grossly, the mass was large, solid, and nodular measuring 7.5×5 ×2.4 cm (Figure 3a). On cut section, creamish white firm areas with necrotic foci were seen. Microscopically, fascicular growth pattern was seen with tumour cells merging with the blood vessel walls. The nuclei were cigar-shaped and blunt-ended with variable atypia, often with cytoplasmic vacuoles at both ends of nuclei (Figures 3b and c). Mitoses were 6-7 /10 HPF. Necrotic foci were present (<50%). IHC, for vimentin and SMA, was positive (Figure 3d), and desmin showed focal faint positivity. CD34 was positive in blood vessels. Based on these findings, a diagnosis of retroperitoneal leiomyosarcoma was made.
DISCUSSION
Leiomyosarcoma is a relatively uncommon tumour with retroperitoneal, primary pulmonary, and cutaneous localisations being even rarer.4,5 The cutaneous leiomyosarcomas arise from the arrectorpili muscle of skin and are common in males aged between 50-70 years, although they may occur at any age.3 Clinically, cutaneous leiomyosarcoma may present with a wide range of differentials including squamous cell carcinoma, amelanotic melanoma, and basal cell carcinoma. The aetiology is unknown, but association has been seen with a preceding history of trauma, irradiation, chemicals, sunlight, and lupus vulgaris. Association with scars has also been seen, most commonly with chronic burn scars, but the pathogenesis of malignant transformation in a burn scar is not known. It has been hypothesised that the scarring damages the lymphatic system, thereby hampering the natural immune surveillance and forming an immunologically privileged site that allows neoplastic changes to occur.6 There are reports of sarcoma developing in a post-burn scar.6 The first case of cutaneous leiomyosarcoma developing in a post-burn scar was reported in 1997 by Can et al. in a 22-year male with a history of burn injury 20 years back.3 They generally have a good prognosis and the risk of metastasis is proportional to the degree of subcutaneous involvement and histological grade with wide surgical excision being the treatment of choice.3 Cutaneous leiomyosarcoma can pose a diagnostic challenge, as was the case with our patient; therefore, any change in an existing scar should be followed by a biopsy in order to make an early diagnosis.
The primary pulmonary sarcomas are rare tumours accounting for 0.5% of all the lung malignancies with leiomyosarcoma constituting 30%.7 They have been classified as intrapulmonary (70%), intraluminal (20%), and pulmonary vascular (10%) depending on their origin from the smooth muscles of the lung parenchyma, bronchial wall, and pulmonary artery, respectively.8 They are mostly found in the 6th decade with a male predominance.9 Clinical presentation may vary or the patient may be asymptomatic with tumour found incidentally on imaging studies.10 Since radiological features are non-specific and the list of differentials is large, histopathological examination is therefore, the gold standard for the diagnosis.11 Differential diagnoses include sarcomatoid carcinoma, monophasic spindle cell synovial sarcoma, malignant peripheral nerve sheath tumour, gastrointestinal stromal tumour, carcinosarcoma, intrapulmonary thymoma, and inflammatory myofibro-blastic tumour.10 Leiomyosarcoma is positive for actin, SMA, desmin, and vimentin.11 Before establishing the diagnosis of primary pulmonary leiomyosarcoma, it is important to rule out metastatic extra pulmonary site. In most of the cases, diagnosis is established after surgical resection, as in our case.10 Prognosis is generally poor and the histological grade of the tumour is generally the most consistent predictor of the long-term survival.10
Retroperitoneal soft-tissue sarcomas comprises 13% of all the adult soft-tissue sarcomas with undifferentiated pleomorphic sarcoma and liposarcomas being the most common.12 About one-half of all the leiomyosarcomas arise in the retroperitoneum involves women being more commonly affected.12 Peak incidence is seen in the seventh decade.12 They are locally invasive large tumours that remain occult for longer periods of time because of a wide expansive area and most cases present with the abdominal discomfort and palpable mass. Gross and microscopic findings are similar to their counterparts elsewhere. The overall 5-year survival rate of retroperitoneal sarcomas is 36%–58% and dependent on tumour histology and extent of tumour invasion.13 Early recognition and aggressive surgery are the keys to long-term survival.
Leiomyosarcomas are uncommon and aggressive malignant tumours arising from the smooth muscle. Because of their clinical and histological overlap with various other neoplasms, they frequently pose diagnostic difficulties. Imaging can be inconclusive, but histological examination is the gold standard for diagnosis. Long-term survival depends on histological grade, extent of invasion, and metastasis with surgical excision remains the mainstay of treatment.
PATIENTS’ CONSENT:
Informed written consent was taken from the patients for publication of the manuscript and photographs.
COMPETING INTEREST:
The authors declared no competing interest.
AUTHORS’ CONTRIBUTION:
BS: Substantial contributions to the conception and design of the work.
SHF: Drafting the work and revising it critically for important intellectual content.
AS: Substantial contributions to the conception and design of the work, acquisition, analysis, and interpretation of data for the work.
JH: Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
All the authors have approved the final version of the manuscript to be published.
REFERENCES
- Massi D, Beltrami G, Mela MM, Pertici M, Capanna R, Franchi A. Prognostic factors in soft tissue leiomyo-sarcoma of the extremities: A retrospective analysis of 42 cases. Eur J Surg Oncol 2004; 30(5):565-72. doi: 10. 1016/j.ejso.2004.03.002.
- Weiss SW, Goldblum JR. Leiomyosarcoma. In: Weiss SW, Goldblum JR, eds. Enzinger and Weiss's Soft Tissue Tumours. 4th ed. Philadelphia, Pa: Mosby Harcourt; 2001: 727-48.
- Can Z, Yilmaz S, Ercocen AR, Apaydin I, Kuzu I. Sarcoma developing in a burn scar: Case report and review of the literature. Burns 1998; 24(1):68-71. doi: 10.1016/ s0305-4179(97)00091-0.
- Shen W, Chen J, Wei S, Wang X, Li X, Zhou Q. Primary pulmonary leiomyosarcoma. J Chin Med Assoc 2014; 77(1):49-51. doi: 10.1016/j.jcma.2013.10.009.
- Chow J, Sabet LM, Clark BL, Coire CI. Cutaneous leiomyosarcoma: Case reports and review of the literature. Ann Plast Surg 1987; 18(4):319-22. doi: 10.1097/0000 0637-198704000-00009.
- Kowal-Vern A, Criswell BK. Burn scar neoplasms: A literature review and statistical analysis. Burns 2005; 31(4):403-13. doi: 10.1016/j.burns.2005.02.015.
- Mastroiani-Etieni B, Falchero L, Chalabreysse L, Chalabreysse L, Loire R, Ranchère D, et al. Primary sarcomas of the lung: A clinic pathologic study of 12 cases. Lung Cancer 2002; 38:283-9. doi: 10.1016/s0169- 5002(02)00303-3.
- Yu HQ, Ren H, Miao Q, Wang Z, Zhang Z, Xu L. Pulmonary leiomyosarcoma. Chin Med Sci J 1997; 12(2):129-31.
- Cordes BG, Collins BT, McDonald JW, Khosla A, Salimi Z. Fine needle aspiration biopsy of primary leiomyosarcoma arising from a pulmonary vein. Acta Cytol 1999; 43(3): 523-6.
- Nath D, Arava S, Joshi P, Madan K, Mathur S. Primary pulmonary leiomyosarcoma of lung: An unusual entity with brief review. Indian J Pathol Microbiol 2015; 58(3):338-40. doi: 10.4103/0377-4929.162868.
- Arnold LM III, Burman SD, O-Yurvati AH. Diagnosis and management of primary pulmonary leiomyosarcoma. J Am Osteopath Assoc 2010; 110(4):244-6.
- Catton CN, O′Sullivan B, Kotwall C, Cummings B, Hao Y, Fornasier V. Outcome and prognosis in retroperitoneal soft tissue sarcoma. Int J Radiat Oncol Biol Phys 1994; 29(5):1005-10. doi: 10.1016/0360-3016(94)90395-6.
- Porter G, Baxter N, Pisters P. Retroperitoneal sarcoma: A population-based analysis of epidemiology, surgery, and radiotherapy. Cancer 2006; 106(7):1610-6. doi: 10.1002/ cncr.21761.