Two Novel Mutations in Ectodysplasin-A Identified in Syndromic Tooth Agenesis
By Weihong Xie, Binghui Zeng, Pei Li, Duoling Xu, Dongsheng Yu, Wei ZhaoAffiliations
doi: 10.29271/jcpsp.2022.05.570ABSTRACT
Objective: To discover novel ectodysplasin‐A (EDA) and wingless‐type MMTV integration site family, member 10A (WNT10A) mutations in tooth agenesis (TA) patients.
Study Design: Case series.
Place and Duration of Study: Guanghua School of Stomatology, Guangzhou, China, from March 2018 to August 2020.
Methodology: EDA and WNT10A were analysed in eleven TA families by PCR and Sanger sequencing. Bioinformatics and structure modelling analyses were performed after identifying different variants, to predict the resulting conformational alterations in WNT10A and EDA.
Results: Two novel mutations (c.796C>A (p.L266I), c.769G>A (p.G257R)) in EDA and two reported mutations (c.637G>A (p.G213S), c.511C>T (p.R171C))in WNT 10A were detected. Combined with the 3D structural analysis, we discovered a correlation between alterations in hydrogen bond formation and the observed phenotypes, potentially affecting protein binding.
Conclusions: The mutations were predicted to be pathogenic through bioinformatics analyses. In addition, by identifying novel mutations, our knowledge regarding the TA spectrum and tooth development was considerably expanded.
Key Words: Anodontia, EDA, WNT 10A, Whole exome sequencing, Odontogenesis.
INTRODUCTION
Congenital tooth agenesis (TA) is a common developmental abnormality. The congenital loss of teeth might cause masticatory, speech, esthetic, and psychological problems, which put a heavy burden on the patients and associated societies. (From preliminary studies, the reported prevalence of this disease ranges from 2.2% to 10.1%.1,2 Whether the patient has other systemic diseases, the agenesis is classified into nonsyndromic tooth agenesis(NSTA) and syndromic tooth agenesis(STA).3
TA can be inherited through an X-linked, autosomal recessive mode or an autosomal dominant trait. Morphogenesis and tooth growth are contingent on the interrelationship between morphogenesis and the epithelium in the course of embryonic development.
During the development of the tooth, over 300 signalling molecules, transcription factors and growth factors are involved in various signalling pathways including the FGF pathway, the SHH pathway, WNT/β-catenin pathway, along with the NF-κB pathway.4,5
Associations have been identified between TA and mutations in ectodysplasin‐A (EDA) and wingless-type MMTV integration site family, member 10A (WNT10A).4,6 Nevertheless, despite records of genomic variants in affected populations, many unidentified genomic mutations causing TA have not been identified to date. Furthermore, the signaling pathways and molecular mechanisms underlying these disorders have not been fully clarified. Therefore, the aim of this study was to discover novel EDA and WNT10A genomic mutations and reveal the genetic basis of TA patients. This expands the TA spectrum and provide a new theoretical basis for the clinical prevention and treatment of TA.
METHODOLOGY
Pakistan with X-linked hypohidrotic ectodermal dysplasia (XLHED). All cases were referred to the Hospital of Stomatology, Sun Yat-Sen University from March 2018 to August 2020 and eleven unrelated Chinese families carrying TA were identified. The absence of permanent teeth was not due to extraction or injuries and was confirmed by all probands. A detailed clinical examination of the systemic and dentition conditions confirmed the diagnosis of NSTA in nine patients and STA in two patients. When needed, dentists, dermatologists, and other doctors thoroughly evaluated the systemic conditions, including the nails, hair, sweat and teeth of the patients.
Criteria with two of the three following systems was considered criteria for XLHED: hypohidrosis, sparse hair, and oligodontia and included.7 Criteria was excluded once the diagnosis of another type of ectodermal dysplasia was found.8
This study was approved by the Ethical Review Committee at the Guanghua School and Hospital of Stomatology at Sun Yat-Sen University. TA patients were recruited, and basic information on TA and its genetic pathogenicity was provided. Related examination and experimental methods were also performed. Their treatment was guaranteed, and convenience was provided if they failed to meet our requirements after the examination. Written informed consent was obtained from the participants, and the recommendations of the Declaration of Helsinki were followed.
From each individual, 4 ml of peripheral blood was collected. Genomic DNA was extracted using the QiaAmp Kit (Qiagen, Düsseldorf, Germany). Applying Oligo 7.0, primers were designed covering the flanking intronic sequences of WNT10A and EDA along with the exons. In previous work, EDA primer sequences were used.9 The products were sequenced using an ABI 3730XL genetic analyser. Sequence Scanner Software v1.0 was utilised to analyse the sequencing results. The nomenclature of mutation, with +1 corresponding to the A of the ATG translation initiation codon of the reference sequences NM_001399.4 (EDA) and NM_025216.2 (WNT10A), was applied.9
Earlier studies described the technique for the bioinformatics analyses performed here.6,9 The Human Protein Reference Database (http://hprd.org/query) provided the domain information for WNT10A and EDA. Applying CLUSTAL X (1.83), the human EDA amino acid sequence (ENST00000374552) was briefly compared to those of the rhesus (ENSMMUT00000024953), cattle (ENSBTAT00000016649), mouse (ENSMUST000001137 79), and chicken (ENSGALT00000007137). PROVEAN, Ploy Phen2, Mutation Taster, and SIFT enabled the prediction of the pathogenic influence of the undiscovered mutations.10-12
Swiss PBD Viewer was used to model the structure of the wild-type and mutant WNT10A and EDA proteins. The structures of WNT-8 (PDB ID 4F0A; X-ray, resolution 3.25 Å) and EDA (PDB ID 1RJ7; X-ray, resolution 2.3 Å) were employed for homology modelling. Figures of three-dimensional structures were built using PyMol v2.4 (The PyMOL Molecular Graphics System, Version 2.4 Schrödinger, LLC., Cambridge, MA, USA).
RESULTS
Congenital absence of teeth excluding other symptoms was observed in three NSTA families, carrying normally shaped teeth. Afflictions other than hypotrichosis, hypohidrosis and hypodontia, were found in two STA patients. Table I shows the numbers of missing permanent teeth. The panoramic radiographs confirmed the diagnosis of TA (Figure 1A–E). Since permanent teeth could have replaced some primary teeth, the numbers of primary teeth of all individuals were not evaluated.
By analysing EDA in XLHED Family 1, we identified one undiscovered mutation c.796C>A (p.Leu266Ile) in the patient and the mother. His mother was an asymptomatic heterozygous carrier of the mutation, while the father was wild-type at c.796. Moreover, a mutation c.637G>A (p.Gly213Ser) was found during our screening of WNT10A. His father carried the mutation with no symptoms and his mother was wild-type at c.637 (Figure 1).
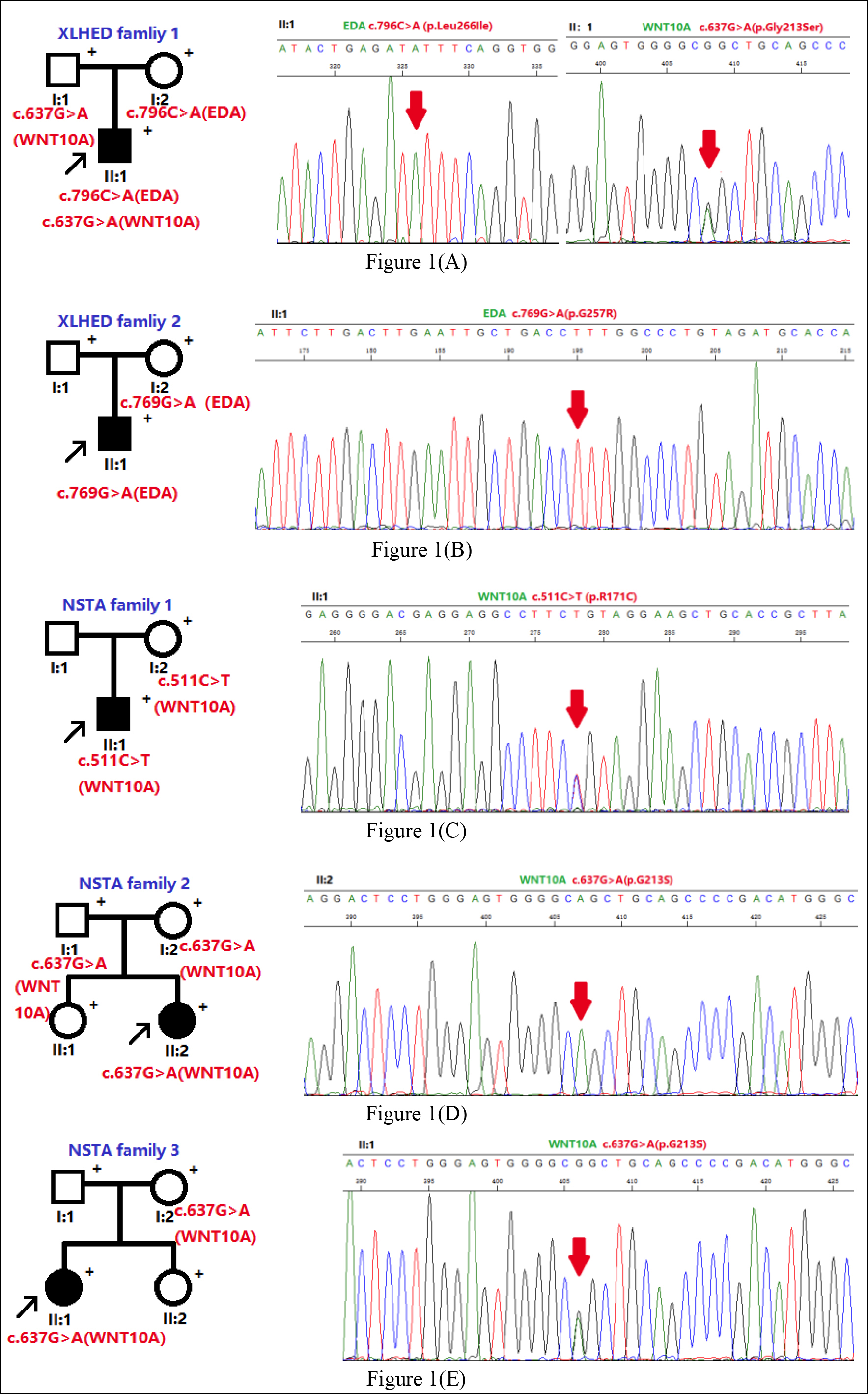 Figure 1: Pedigrees and corresponding mutation of XLHED Families 1 and 2 and NSTA Families 1–3. (A) In XLHED Family 1, an EDA mutation c.796C>A (p.Leu266Ile) was discovered, and a known WNT10A mutation c.637G>A(p.Gly213Ser) was found. (B) One novel EDA mutation c.769G>A (p.Gly257Arg) was discovered in XLHED Family 1. (C) In NSTA Family 1, the patient had the heterozygous WNT10A mutation c.511C>T (p.Arg171Cys). (D)Moreover, for NSTA Family 2, one covered homogeneous WNT10A mutation c.637G>A (p.Gly213Ser) was confirmed within the gene of the patient. (E) For NSTA Family 3, the patient was confirmed to carry the WNT10A mutation c.637G>A (p.Gly213Ser)). Black arrows indicate the probands. Each individual for whom blood samples were obtained is indicated with ‘+’. Arrows indicate the mutations. XLHED: X‐linked hypohidrotic ectodermal dysplasia. NSTA: nonsyndromic tooth agenesis.
Figure 1: Pedigrees and corresponding mutation of XLHED Families 1 and 2 and NSTA Families 1–3. (A) In XLHED Family 1, an EDA mutation c.796C>A (p.Leu266Ile) was discovered, and a known WNT10A mutation c.637G>A(p.Gly213Ser) was found. (B) One novel EDA mutation c.769G>A (p.Gly257Arg) was discovered in XLHED Family 1. (C) In NSTA Family 1, the patient had the heterozygous WNT10A mutation c.511C>T (p.Arg171Cys). (D)Moreover, for NSTA Family 2, one covered homogeneous WNT10A mutation c.637G>A (p.Gly213Ser) was confirmed within the gene of the patient. (E) For NSTA Family 3, the patient was confirmed to carry the WNT10A mutation c.637G>A (p.Gly213Ser)). Black arrows indicate the probands. Each individual for whom blood samples were obtained is indicated with ‘+’. Arrows indicate the mutations. XLHED: X‐linked hypohidrotic ectodermal dysplasia. NSTA: nonsyndromic tooth agenesis.
Table I: Clinical data and corresponding mutations of EDA and WNT10A genes.
|
Family Number |
Proband |
Gender & Age |
Corresponding gene |
Nucleotide Varients |
Genotype |
Predicted Protein Changes |
Number of absent Teeth # |
|
XLHED family 1 |
II:1 |
Male,4Y |
EDA |
c.796G>A |
Hemizygous |
p.Leu266Ile |
22 |
|
WNT10A |
c.637C>T |
Heterozygous |
p.Gly213Ser |
||||
|
XLHED family 2 |
II:1 |
Male,8Y |
EDA |
c.769C>A |
Hemizygous |
p.Gly257Arg |
21 |
|
NSTA family 1 |
II:1 |
Male,12Y |
WNT10A |
c.511C>T |
Heterozygous |
p.Arg171Cys |
2 |
|
NSTA family2 |
II:2 |
Female,7Y |
WNT10A |
c.637G>A |
Homozygous |
p.Gly213Ser |
11 |
|
NSTA Family3 |
II:1 |
Female,7Y |
WNT10A |
c.637G>A |
Heterozygous |
p.Gly213Ser |
7 |
|
#The third molars and deciduous teeth were not included. Bold type: undiscovered mutation; Genotype: Homozygous, Heterozygous or Hemizygous. XLHED: X‐linked hypohidrotic ectodermal dysplasia. NSTA: nonsyndromic tooth agenesis. |
|||||||
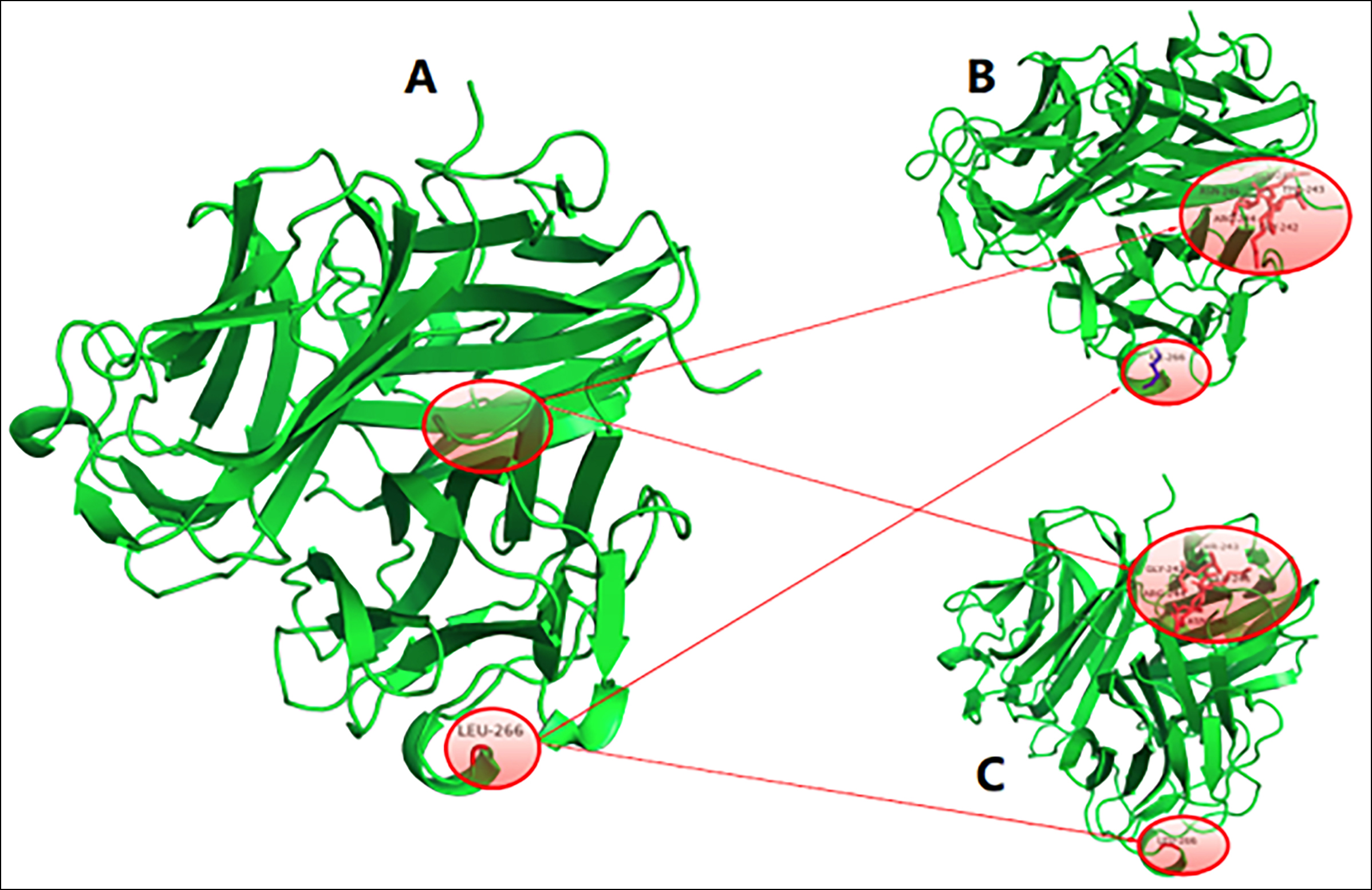 Figure 2: Tumour necrosis factor homology domain of wild-type EDA protein (2-A), Leu266Ile EDA protein (2-B) and Gly257Arg EDA protein (2-C). The three trimers were similar in morphology and structure but slightly different in primary structures. There were five more amino acid residues in the Leu266Ile EDA protein (B) and Gly257Arg EDA protein (2-C) than in the wild-type EDA protein (2-A): GLY242, THR243, ARG244, GLU245 and ASN246. These five amino acids had close hydrogen bond interactions with the surrounding residues. In addition, in the Leu266Ile EDA protein (2-B), the original LEU266 was replaced by ILE.
Figure 2: Tumour necrosis factor homology domain of wild-type EDA protein (2-A), Leu266Ile EDA protein (2-B) and Gly257Arg EDA protein (2-C). The three trimers were similar in morphology and structure but slightly different in primary structures. There were five more amino acid residues in the Leu266Ile EDA protein (B) and Gly257Arg EDA protein (2-C) than in the wild-type EDA protein (2-A): GLY242, THR243, ARG244, GLU245 and ASN246. These five amino acids had close hydrogen bond interactions with the surrounding residues. In addition, in the Leu266Ile EDA protein (2-B), the original LEU266 was replaced by ILE.
By sequencing the EDA genes in the XLHED Family 2, we uncovered a new mutation c.769G>A (p.Gly257Arg), shared by the proband and his mother. His father was wild-type at c.769.
Both EDA and WNT10A genes were examined in the patients of three NSTA families. A WNT10A mutation c.511C>T (p.Arg171Cys) was discovered within NSTA Family 1. His mother carried the mutation with no symptoms. Blood specimens of the father were not available. For Family 2, the covered homozygous WNT10A mutation c.637G>A (p.Gly213Ser) was found in the genome of the proband. Both the father and the mother were heterozygous at c.637, manifesting no symptoms. Within NSTA Family 3, we discovered the covered WNT10A mutation c.637G>A (p.Gly213Ser) in the proband as well as her mother (Figure 1).
As mentioned before, EDA variants c.796C>A were discovered in XLHED Family 1 and c.769G>A in XLHED Family 2. Bioinformatic analyses and experiments were performed to determine whether they were pathogenic.
The EDA mutation c.796C>A (p.Leu266Ile) found in XLHED Family 1 was not published in Pubmed, the Human Gene Mutation Database (HGMD; public version) or 1000 Genomes. A leucine was substituted for an isoleucine because of the variant at the position. It was by PolyPhen2 predicted to be ‘possibly damaging’. Moreover, the prediction of Mutation Taster was ‘disease-causing’ since the mutation transferred the amino acid sequence and changed the splice site.
Another mutation, c.769G>A (p.Gly257Arg), was also unavailable in Pubmed, the Human Gene Mutation Database or 1000 Genomes. A glycine was replaced with an arginine caused by the variant at the position. PolyPhen2 predicted it as being ‘probably damaging’. A status of ‘disease-causing’ was predicted by Mutation Taster because it altered the amino acid sequence. Moreover, the mutation may obstruct the link between the 2nd and 3rd exon areas, resulting in protein features changes.
Structural modelling of the tumour necrosis factor homology domain of the wild-type EDA protein, Leu266Ile EDA protein and Gly257Arg EDA protein (Figure 2) indicated similar morphologies and structures but slightly different primary structures. There were five more amino acid residues in the Leu266Ile EDA protein (Figure 2-B) and Gly257Arg EDA protein (Figure 2-C) than in the wild-type EDA protein (Figure 2-A): GLY242, THR243, ARG244, GLU245 and ASN246. These five amino acids had close hydrogen bond interactions with the surrounding residues. In addition, in the Leu266Ile EDA protein, the original LEU266 was replaced by ILE.
DISCUSSION
This study presents synthetic genetic research on NSTA and XLHED. Genetic mutations in five families carrying NSTA or XLHED were discovered along with Sanger sequencing. EDA mutations c.796C>A (p.Leu266Ile) as well as c.769G>A (p.Gly257Arg) were novel. Structural modelling and bioinformatics analyses were performed to analyse the pathogenicity of the mutations.
Notably, in the XLHED Family 1, simultaneous EDA and WNT10A mutations resulted in STA. In the XLHED Family 1, the novel mutation c.796C>A (p.Leu266Ile) was present in the patient along with the mother. In addition, a previously identified mutation c.637G>A (p.Gly213Ser) was found in the patient and his father through our screening of WNT10A. Both parents were asymptomatic. In previous research, the WNT10A mutation c.637G>A (p.Gly213Ser) was identified and analysed, and it was considered to contribute to TA.13 Therefore, whether the newly discovered mutation can lead to XLHED is worthy of detailed discussion. Conversely, the corresponding mutation is the most significant pathogenic factor in XLHED, since EDA results in more than half of TA cases.14 Moreover, according to the bioinformatics analyses, including cross-species comparison, PolyPhen2, and structural modelling, c.796C>A (p.Leu266Ile) is likely to be pathogenic. The phenomenon in which the mother had no symptoms could be explained by female carriers of EDA mutations presenting with a normal or very mild phenotype, since the EDA gene is located on chromosome X.15,16 In contrast, the phenotype of the patient seemed more severe than the previous single WNT10A mutation c.637G>A (p.Gly213Ser) patient reported previously.13 Clinical manifestations further support this conjecture. Previous studies have indicated that mutations in different genes could also lead to one disease in one patient.17,18
EDA and WNT10A mutations can influence each other resulting in TA.19 In this patient, both EDA and WNT10A mutations were found, and as mentioned earlier in the article, analyses indicated that the EDA and WNT10A mutations may affect protein function. Therefore, it was conjectured that the interaction between EDA and WNT10A mutations might result in XLHED.
The same WNT10A mutation c.637G>A(p.Gly213Ser) was found in XLHED Family 1, NSTA family2 and NSTA family 3. In NSTA family 2, the proband was homozygous and showed TA. Both the father and the mother were heterozygous at c.637, showing no symptoms.
In XLHED Family 1, the proband and his father carried the same mutation gene, and were both heterozygous. While the proband showed TA, his father was asymptomatic. This phenomenon also occurred in the proband and her mother in NSTA Family 3. This genetic phenomenon might be explained as irregular dominance and was reported in TA before.19,20 Other gene factors may be involved in regulating WNT10A c.637 gene expression during tooth development. Individuals with different genetic backgrounds could be an important cause of irregular dominant inheritance. Environmental factors might also be one of the reasons for the irregular dominance. Nevertheless, further research is needed in the future to reveal the mechanism in a larger sample.
The proband and his mother shared another new EDA mutation c.769G>A (p.Gly257Arg) in XLHED Family 2. His father was wild-type at c.769. For EDA mutation c.769G>A, a glycine was replaced with an arginine, caused by the mutation at the position. In the previous report, the same site but different nucleotide mutation of EDA c.769G>C(p.G257R) was found in the patient with NSTA.21 It is speculated that tooth development at this site is more sensitive to the mutation of EDA, providing a more sufficient basis for c.769G>A to be a pathogenic mutation. This can be explained as the mutation of EDA at c.769, affecting the protein and playing an important role in tooth development. PolyPhen2 and Mutation Taster predicted it to be disease-causing as it altered the amino acid sequence. This conclusion is also supported by the comparative structural modelling of the EDA protein.
In recent years, there have been many reports about novel mutations involved EDA-EDAR-NF-κB and WNT/beta-catenin signaling pathways in TA.22,23 A close relationship is likely to exist between EDA and WNT pathways. Previous studies showed that the WNT and EDA pathways had a sequential interdependency during hair follicle morphogenesis and development.24 Moreover, a cascade of WNT and EDA signaling was reported to be essential for touch dome Merkel cell development.25 Therefore, there might be an interaction between EDA and WNT10A mutations, which together result in abnormalities in odontogenesis.
CONCLUSIONS
This research confirmed two novel EDA mutations c.769G>A (p.Gly257Arg) and c.796C>A (p.Leu266Ile) and two discovered WNT10A mutations c.511C>T (p.Arg171Cys) and c.637G>A (p.Gly213Ser) among eleven TA patients. This study extrapolated the two undiscovered mutations as pathogenic representing a contribution to precision medicine.
ETHICAL APPROVAL:
This study was approved by the Ethical Review Committee at the Guanghua School and Hospital of Stomatology at Sun Yat-Sen University.
PATIENT’S CONSENT:
Informed consent was obtained from patients to publish the data concerning this case.
COMPETING INTERESTS:
The authors declared no conflict of interest.
ACKNOWLEDGEMENTS:
This project was funded by the National Natural Sciences Foundation of China (81873711 and 81974146) and the Science and the Fundamental Research Funds of the Central Universities, Sun Yat-sen University (19ykpy86), and China Postdoctoral Science Foundation (2020M673023).
AUTHORS’ CONTRIBUTION:
WX: Conceptualisation, investigation, methodology, writing-original draft, and writing-review and editing.
BZ: Data curation, investigation, validation, and writing-original draft.
Pei Li: Data curation, and methodology.
Duoling Xu: Data curation.
DY: Data curation, investigation, methodology, writing-original draft, and writing-review and editing.
Wei Zhao: Conceptualisation, funding acquisition, supervision, writing-original draft, writing-review and editing.
All authors approved the final version of the manuscript to be published.
REFERENCES
- Polder BJ, Van't Hof MA, Van der Linden FP, Kuijpers-Jagtman AM. A meta-analysis of the prevalence of dental agenesis of permanent teeth. Community Dent Oral Epidemiol 2004; 32(3):217-26. doi: 10.1111/j.1600- 0528.2004.00158.x.
- Zhang J, Liu HC, Lyu X, Shen GH, Deng XX, Li WR, et al. Prevalence of tooth agenesis in adolescent Chinese populations with or without orthodontics. Chinese J Dent Res 2015; 18(1):59-65. doi: 10.3290/j.cjdr.a33969.
- Al-Ani AH, Antoun JS, Thomson WM, Merriman TR, Farella M. Hypodontia: An update on its etiology, classification, and clinical management. BioMed Res Int 2017; 2017:9378325. doi: 10.1155/2017/9378325.
- Yu M, Wong SW, Han D, Cai T. Genetic analysis: Wnt and other pathways in nonsyndromic tooth agenesis. Oral Diseases 2019; 25(3):646-51. doi: 10.1111/odi.12931.
- Li J, Parada C, Chai Y. Cellular and molecular mechanisms of tooth root development. Development 2017; 144(3):374-84. doi: 10.1242/dev.137216.
- Zeng B, Lu H, Xiao X, Zhou L, Lu J, Zhu L, et al. Novel EDA mutation in X-linked hypohidrotic ectodermal dysplasia and genotype-phenotype correlation. Oral Disea 2015; 21(8): 994-1000. doi: 10.1111/odi.12376.
- Gupta AA, Gotmare SS, Jain M, Pereira T, Khare P. Hypohydrotic ectodermal dysplasia in an indian family. J Coll Physi Surg Pak 2019; 29(4):381-3. doi: 10.29271/jcpsp.2019. 04.381.
- Reyes-Reali J, Mendoza-Ramos MI, Garrido-Guerrero E, Méndez-Catalá CF, Méndez-Cruz AR, Pozo-Molina G. Hypohidrotic ectodermal dysplasia: Clinical and molecular review. Int J Dermatol 2018; 57(8):965-72. doi: 10.1111/ijd.14048.
- Zeng B, Zhao Q, Li S, Lu H, Lu J, Ma L, et al. Novel EDA or EDAR mutations identified in patients with X-linked hypohidrotic ectodermal dysplasia or non-syndromic tooth agenesis. Genes 2017; 8(10):259. doi: 10.3390/genes8100259.
- Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet 2016; 99(4):877-85. doi: 10.1016/j.ajhg.2016. 08.016.
- Ning J, Ahmed S, Cheng G, Chen T, Wang Y, Peng D, et al. Analysis of the stability and affinity of BlaR-CTD protein to β-lactam antibiotics based on docking and mutagenesis studies. J Biol Engin 2019; 13:27. doi: 10.1186/s13036- 019-0157-4.
- Li R, Xu Z, Zhao D, Zhang Y, Xie Z, Wang C, et al. Analysis of MCCC2 gene variant in a pedigree affected with 3-methylcrotonyl coenzyme A carboxylase deficiency. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2021; 38(1):74-7. doi: 10.3760/cma.j.cn511374-20200110-00021.
- Plaisancié J, Bailleul-Forestier I, Gaston V, Vaysse F, Lacombe D, Holder-Espinasse M, et al. Mutations in WNT10A are frequently involved in oligodontia associated with minor signs of ectodermal dysplasia. Am J Med Genetics Part A 2013; 161a(4):671-8. doi: 10.1002/ajmg.a.35747.
- Cluzeau C, Hadj-Rabia S, Jambou M, Mansour S, Guigue P, Masmoudi S, et al. Only four genes (EDA1, EDAR, EDARADD, and WNT10A) account for 90% of hypohidrotic/anhidrotic ectodermal dysplasia cases. Human Mutation 2011; 32(1):70-2. doi: 10.1002/humu.21384.
- Nakata M, Koshiba H, Eto K, Nance WE. A genetic study of anodontia in X-linked hypohidrotic ectodermal dysplasia. Am J Hum Genet 1980; 32(6):908-19.
- Lexner MO, Bardow A, Juncker I, Jensen LG, Almer L, Kreiborg S, et al. X-linked hypohidrotic ectodermal dysplasia. Genetic and dental findings in 67 Danish patients from 19 families. Clinical Genetics 2008; 74(3):252-9. doi: 10.1111/j.1399- 0004.2008.01037.x.
- Badiner N, Taylor SP, Forlenza K, Lachman RS, Bamshad M, Nickerson D, et al. Mutations in DYNC2H1, the cytoplasmic dynein 2, heavy chain 1 motor protein gene, cause short-rib polydactyly type I, Saldino-Noonan type. Clin Genetics 2017; 92(2):158-65. doi: 10.1111/cge.12947.
- Lejeune S, Guillemot F, Triboulet JP, Cattan S, Mouton C, Porchet N, et al. Low frequency of AXIN2 mutations and high frequency of MUTYH mutations in patients with multiple polyposis. Human Mutation 2006; 27(10):1064. doi: 10.1002/humu.9460.
- Zhao K, Lian M, Zou D, Huang W, Zhou W, Shen Y, et al. Novel mutations identified in patients with tooth agenesis by whole-exome sequencing. Oral Diseases 2019; 25(2):523-34. doi: 10.1111/odi.13002.
- Zeng B, Xiao X, Li S, Lu H, Lu J, Zhu L, et al. Eight mutations of three genes (EDA, EDAR, and WNT10A) Identified in seven hypohidrotic ectodermal dysplasia patients. Genes 2016; 7(9). doi: 10.3390/genes7090065.
- He HY, Liu Y, Han D, Liu HC, Bai BJ, Feng HL. [EDA mutation screening and phenotype analysis in patients with tooth agenesis]. Beijing Da Xue Xue Bao Yi Xue Ban J Peking University Health Sci 2016; 48(1):686-91. doi: 10.3969/j.issn.1671- 167X.2016.04.024.
- Zhang H, Kong X, Ren J, Yuan S, Liu C, Hou Y, et al. A novel EDAR missense mutation identified by whole-exome sequencing with non-syndromic tooth agenesis in a Chinese family. Molecular Genetics Genomic Medi 2021; 9(6):e1684. doi: 10.1002/mgg3.1684.
- Park H, Song JS, Shin TJ, Hyun HK, Kim YJ, Kim JW. WNT10A mutations causing oligodontia. Archives Oral Biology 2019;103:8-11. doi: 10.1016/j.archoralbio.2019.05.007.
- Zhang Y, Tomann P, Andl T, Gallant NM, Huelsken J, Jerchow B, et al. Reciprocal requirements for EDA/EDAR/NF-kappaB and Wnt/beta-catenin signaling pathways in hair follicle induction. Developmental Cell 2009; 17(1):49-61. doi: 10.1016/j.devcel.2009.05.011.
- Xiao Y, Thoresen DT, Miao L, Williams JS, Wang C, Atit RP, et al. A cascade of Wnt, Eda, and Shh signaling is essential for touch dome merkel cell development. PLoS Genetics 2016; 12(7):e1006150. doi: 10.1371/journal.pgen.1006150.