Takotsubo Like Cardiomyopathy with Concomitant Pheochromocytoma: A Rare Presentation
By Pirbhat Shams1, Intisar Ahmed1, Sabeeh Siddique2, Farhat Abbas1, Aamir Hameed Khan1Affiliations
doi: 10.29271/jcpsp.2022.11.1483ABSTRACT
Pheochromocytoma classically presents with headache, diaphoresis, palpitations and, raised blood pressure. Rarely, it manifests as cardiomyopathy. Herein, we present a case of a 42-year woman who presented with heart failure and on work-up was found to have pheochromocytoma leading to Takotsubo-like cardiomyopathy. The biochemical profile revealed raised serum metanephrines and normetanephrines. CT abdomen showed a left adrenal mass. Within two weeks of presentation and before surgical excision of the mass, she recovered from cardiomyopathy. After medical optimisation, the patient underwent elective adrenalectomy, which on histological evaluation revealed pheochromocytoma.
Key Words: Cardiomyopathy, Pheochromocytoma, Adrenal mass.
INTRODUCTION
Pheochromocytoma is a rare catecholamine secreting tumour arising from chromaffin cells of adrenal medulla. It carries an annual incidence of 0.8 per 100,000 person-years1 and classically presents with headache, diaphoresis, tachycardia, and raised blood pressure. Takotsubo cardiomyopathy is a syndrome comprising of stress cardiomyopathy, transient cardiac segmental wall motion abnormalities or left ventricular dysfunction or ST-T changes in association with normal coronary arteries. There is growing evidence of pheochromocytoma and associated tumours presenting with cardiovascular manifestations such as dilated or hypertrophic obstructive cardiomyopathy, arrhythmias, cardiogenic shock, and less commonly as Takotsubo-like cardiomyopathy (TLC). The catecholamine-induced cardiac toxicity is not only attributable to its direct toxic effect on the myocardium, but also via receptor mediated cardiac stunning.2 Here, we present a case of a 42-year- woman presenting with heart failure who was found to have pheochromocytoma.
CASE REPORT
A 42-year- woman, with recently diagnosed hypertension, presented to the emergency department with complaints of abdominal pain, palpitation, and headache for 10 days.
Abdominal pain was localised to the epigastrium and radiated to the back and both shoulders. It was associated with several episodes of projectile vomiting, palpitations, headache, and vertigo. She also presented with sudden onset shortness of breath and trepidation for last the 3 days. There was history of persistently raised blood pressures inadequately controlled by oral medications. She reported chronic constipation for the last 10 years. No emotional or physical stressors were identified. There was no history of fever, seizures or blurring of vision. Of note, there was no family history of cardiovascular or renal diseases. On presentation, she was hypertensive (200/110 mmHg), tachycardiac (120 beats per minute), and tachypneic (31 breaths per minute). She had oxygen saturation of 92% on room air. Examination revealed bibasilar fine crackles in chest and raised jugular venous pulse. Neurological and abdominal examinations were unremarkable.
A set of blood investigations was done which showed Haemoglobin of 13.5 g/dl, arterial lactate of 6.7 mmol/L, White blood cell count of 25×109 /L, troponin of 12 ng/ml, repeat troponin of 19 ng/ml, and NT-ProBNP of 12600 pg/ml. Liver function tests, serum lipase, and renal function tests were normal. Echocardiogram was done which showed ejection fraction of 20% and segmental akinesia. Basal septal and basal inferior segments were contracting normally while the rest of the segments were akinetic with grade II diastolic dysfunction. Based on the presentation with acute abdominal pain and history of chronic constipation, the possibility of mesenteric ischemia was considered, and arterial lactate level was sent which was found to be elevated. Electrocardiogram showed sinus tachycardia with no specific ST-T changes. Chest X-ray was consistent with pulmonary oedema.
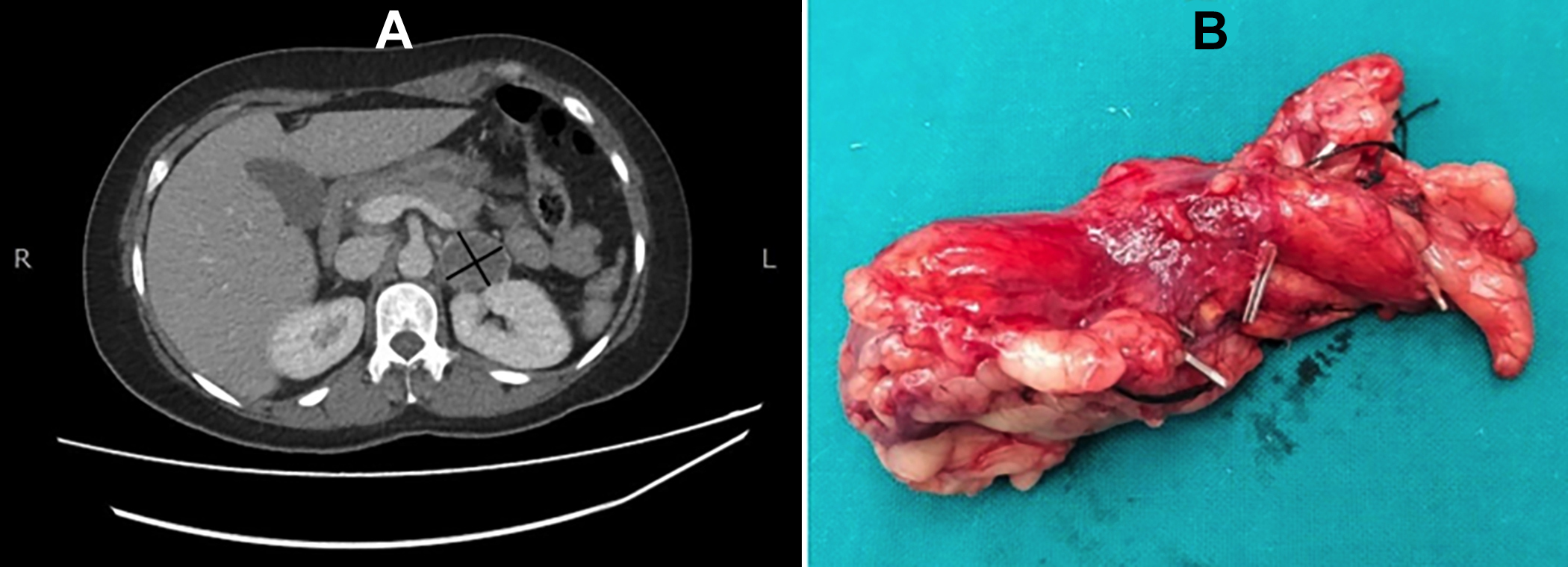 Figure 1: CT abdomen with contrast showing a well-circumscribed, rounded, soft tissue density lesion identified in the left adrenal gland (A). Left adrenal mass after adrenalectomy (B).
Figure 1: CT abdomen with contrast showing a well-circumscribed, rounded, soft tissue density lesion identified in the left adrenal gland (A). Left adrenal mass after adrenalectomy (B).
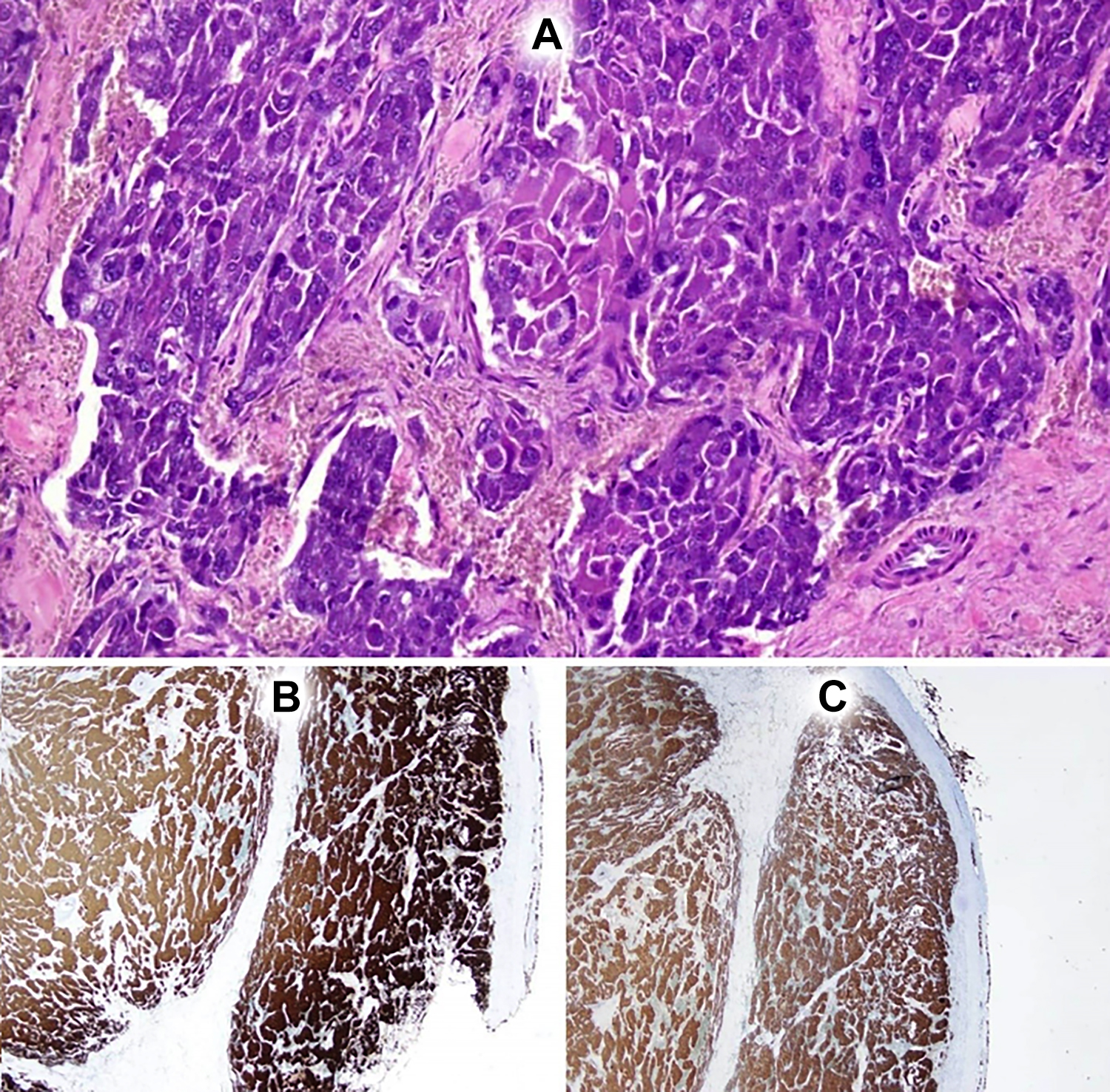 Figure 2: Adrenal mass lesion exhibiting a medullary neoplastic lesion in trabecular and nested pattern. Individual cells are arranged in Zellballen pattern (A). Synaptophysin stain was positive (B). Chromogranin stain was positive (C).
Figure 2: Adrenal mass lesion exhibiting a medullary neoplastic lesion in trabecular and nested pattern. Individual cells are arranged in Zellballen pattern (A). Synaptophysin stain was positive (B). Chromogranin stain was positive (C).
Because of abdominal pain and raised lactate levels, CT abdomen with contrast was done (Figure 1) to rule out mesenteric ischemia and CT angiogram aorta was done to rule out possible aortic dissection. The patient was admitted to the coronary care unit with an impression of non-ST elevation acute coronary syndrome. She underwent a coronary angiogram within 48 hours of admission which showed normal coronary arteries. Meanwhile, CT abdomen reported no mesenteric ischemia or aortic dissection, a normal pancreas but incidental lesion in the left adrenal gland with peripheral enhancement. Based on the finding of adrenal mass and clinical presentation of hypertension, the following set of biochemistry was ordered: Serum metanephrines of 553 pg/ml (Normal: 0-90 pg/ml), Serum normetanephrine of 1591 pg/ml (Normal: 0-190 pg/ml) and 24-hour urinary VMA levels of 6 mg in 2400 ml urine/24 hours (Normal: <13 mg). A diagnosis of pheochromocytoma-induced TLC (pheo-TLC) was made. The patient was managed with intravenous diuretics and non-invasive ventilation and she subsequently improved. She was started on alpha blockers during hospital stay on which her blood pressure normalised. She was thereafter discharged and beta-blocker was started due to subsequent clinic visits. A repeat echocardiogram in two weeks revealed normal left ventricular function and no segmental wall motion abnormalities. Of note, the cardiomyopathy reversed to normal even before the surgical excision of pheochromocytoma. She was then re-admitted for elective left adrenalectomy and had an uneventful perioperative course except for transient fluid-responsive drop in mean arterial pressure in the immediate postoperative period. Histopathology of the excised mass confirmed pheochromocytoma (Figure 2). The patient was followed up regularly and she showed improved NYHA functional class at two weeks. A repeat echocardiogram was done which showed an ejection fraction of 60%. No recurrent episodes of hypertension or vomiting were observed.
DISCUSSION
The revised taskforce consensus on TLC has identified pheochromocytoma as a possible trigger for TLC.3 This is in contrast to the original Mayo clinic criteria which required the absence of pheochromocytoma as a criteria for diagnosing TLC.4 TLC is being increasingly recognised as a presentation for catecholamine-secreting tumours.5-7 In an analysis of 80 published cases of pheo-TLC worldwide, comparison was made with all cause TLC and following were the salient features: female predominance, relatively younger age at presentation, basal segments’ involvement in 30%, global involvement in 20%, higher complication rate and recurrence rate than all cause TLC and no mortality difference between the two groups. Age less than 50 years and global and basal segmental wall motion abnormalities were identified as risk factors for complications.8 There are several case reports of pheo-TLC worldwide and the western world seems to be the major contributor.5-7 However, there is scarcity of such case reports from South Asia region, indicating that some geographical, racial, or environmental factors modify the occurrence and presentation of pheo-TLC.9,10
Moreover, pheo-TLC cases worldwide show that reversal of cardiomyopathy usually occurs after surgical correction. Interestingly, this patient recovered from cardiomyopathy even before the surgical excision of pheochromocytoma, hence mimicking stress cardiomyopathy. TLC remains underdiagnosed. It is known to follow stress of any sort leading to the increased catecholaminergic release in the body. However, before attributing all cases to emotional or physical stress, we think it is prudent to look for underlying organic cause such as catecholamine-secreting tumours, particularly, when there is absence of identifiable physical or emotional stressor, as in this case. Moreover, even in the presence of ischemic heart disease, tumours should still be considered as differentials in anyone presenting with recurrent heart failure after revascularisation.
PATIENT’S CONSENT:
Informed consent was obtained from patients to publish the data concerning this case.
COMPETING INTEREST:
The authors declared no competing interest.
AUTHORS’ CONTRIBUTION:
PS, IA: Composed the manuscript.
SS, FA: Reviewed it.
AHK: Conception and supervised manuscript.
All the authors have approved the final version of the manuscript to be published.
REFERENCES
- Sutton MG, Sheps SG, Lie JT. Prevalence of clinically unsuspected pheochromocytoma. Review of a 50-year autopsy series. Mayo Clinic Proceedings 1981; 56(6): 354-60.
- Kassim TA, Clarke DD, Mai VQ, Clyde PW, Mohamed Shakir KM. Catecholamine-induced cardiomyopathy. Endocrine Pract 2008; 14(9):1137-49. doi: 10.4158/EP. 14.9.1137.
- Ghadri J-R, Wittstein IS, Prasad A, Sharkey S, Dote K, Akashi YJ, et al. International expert consensus document on takotsubo syndrome (Part I): Clinical characteristics, diagnostic criteria, and pathophysiology. Eur Heart J 2018; 39(22):2032-46. doi: 10.1093/eurheartj/ehy076.
- Scantlebury DC, Prasad A. Diagnosis of Takotsubo cardiomyopathy. Circ J 2014; 78(9):2129-39. doi: 10. 1253/circj.cj-14-0859.
- Diaz B, Elkbuli A, Ehrhardt JD, McKenney M, Boneva D, Hai S. Pheochromocytoma-related cardiomyopathy presenting as broken heart syndrome: Case report and literature review. Int J Surg Case Rep 2019; 55:7-10. doi: 10.1016/j.ijscr.2018.12.003.
- Butt K, Ali S, Sattar Z, Ur Rahman A, Burt JR. Funny lumps, flaming pheo, and a broken heart: A rare case of pheochromocytoma. Cureus 2018; 10(11):e3646. doi: 10.7759/cureus.3646.
- Contreras Gutiérrez VH. Takotsubo cardiomyopathy: A case-report. Revista Médica del Hospital General de México 2018; 81:41-6.
- Hassan SY. Clinical features and outcome of pheochromocytoma-induced takotsubo syndrome: Analysis of 80 published cases. Am J Cardiol 2016; 117(11):1836-44. doi: 10.1016/j.amjcard.2016.03.019.
- Varghese RT, John AM, Paul TV. Catecholamine induced cardiomyopathy in pheochromocytoma. Indian J Endocrinol Metab 2013; 17(4):733-5. doi: 10.4103/2230- 8210.113771.
- Chungsomprasong P, Hamilton R, Luining W, Fatah M, Yoo SJ, Grosse-Wortmann L. Left ventricular function in children and adolescents with arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol 2017; 119(5): 778-84. doi: 10.1016/j.amjcard.2016.11.020.