Spectrum of Cystic Fibrosis Conductance Regulator Gene Mutations Reported in Pakistani Descent Cystic Fibrosis Patients
By Hafsa Majid, Aysha Habib Khan, Syed Bilal Hashmi, Tariq Moatter, Asghar NasirAffiliations
doi: 10.29271/jcpsp.2022.08.1042ABSTRACT
Cystic fibrosis (CF) is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. This study aims to determine the genotypic and phenotypic spectrum of the CFTR gene mutations reported in the literature for Pakistani-origin CF patients. Databases were searched for such studies from 1947-2019 for sample size, method of diagnosis, and CFTR gene mutations. The authors identified 12 studies reporting 33 CFTR gene mutations, both intronic as well as exonic in Pakistani origin patients. The most widely tested mutation was D508 with a frequency of 17%-60%. No hotspot zone was identified and not all reported mutations were causing disease. There is a need to identify common mutations in the Pakistani population to develop population-specific CFTR mutations panel. This will enable the researchers to perform phenotype-genotype correlation studies to improve the CF detection rate.
Key Words: Cystic fibrosis, Pakistan, Mutations, CFTR.
INTRODUCTION
Cystic fibrosis (CF) is a potentially life limiting recessive genetic disease, affecting 0.737/10,000 live births in European countries.1 Approximately 2,500 babies are born with CF each year in the United States. About 1 in every 20 Americans is unaffected carrier of an abnormal CF gene. It is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene that encodes the CFTR protein which participates in fluid homeostasis across mucosal surfaces.2 Optimal outcome of the CF patient depends on early diagnosis. According to the Cystic Fibrosis Foundation consensus report, the criteria for the diagnosis of CF is: a) evidence of a CFTR abnormality together with biological evidence of channel dysfunction (i.e., abnormal sweat chloride concentration) or b) identification of a CF disease-causing mutation in CFTR gene.3,4
Multiple case reports have been published from Pakistan’s population, but no study has reported incidence or prevalence. The incidence of CF in Pakistani-descent immigrants residing in the United Kingdom is 1 in 10,800 to 12,000.5,6
The exact incidence in Pakistan is unknown, however, it is expected to be high considering the high cousin marriage rate.7 It is considered an underdiagnosed disease in Pakistan owing to the lack of diagnostics for confirmation of CF in this country. Most of the patients reported are diagnosed based on suggestive clinical features.
The CFTR gene is composed of 27 exons. Until now, more than 2,000 mutations in the CFTR gene have been reported.8 The CF disease phenotype varies with the type and number of mutations present. Not all the mutations identified are associated with CF but can be present in related disorders ranging from sinusitis to severe lung, pancreatic, or liver disease.3 Genotype is reported to be associated with the severity of disease, symptoms only occur in the presence of combination of specific mutations which in turn affect the management and prognosis of the patient. Patients presenting with pancreatic insufficiency have different haplotypes compared to the pancreatic-sufficient patients.9 That is why the identification of mutations in Pakistani population and their frequencies is critically important for understanding CF disease dynamics and the management of the patients.
The nature, distribution, and frequency of CFTR mutations depend on the population studied and the method employed. PCR is considered less sensitive compared to sequencing for the identification of mutations.3,10 In this study, the authors systematically reviewed the studies reporting CFTR gene mutations in Pakistani-origin patients with CF. The aim of the review was to determine the genotypic and phenotypic spectrum of CFTR gene mutations reported in Pakistani origin patients.
METHODOLOGY
Both retrospective and prospective studies about CFTR gene mutations in Pakistani or Pakistani origin patients with CF were included from 1947 to 2019. Abstracts and full-text studies both were included. Review and non-English language articles were excluded. The PubMed and PakMedinet databases were independently searched by two reviewers (HM and SBH).
Search terms were “Cystic Fibrosis” and “Pakistan” and “mutation” as MeSH major topics in the PubMed database. In PakMedinet data base, the terms “Cystic Fibrosis” and “mutation” were looked for in abstracts.
The reviewers selected studies with relevant titles only. Duplicate studies were also excluded from the present study. Abstracts of all the relevantly titled studies were studied and studies reporting CFTR gene mutations in Pakistani or Pakistani origin patients with CF were included. Full text of all the studies was read and data was extracted from it. The data regarding the year of study, sample size of the cases, method of CF diagnosis, and CFTR gene mutations report were included. The process of inclusion of studies is detailed as a `CONSORT DIAGRAM` in Figure 1.
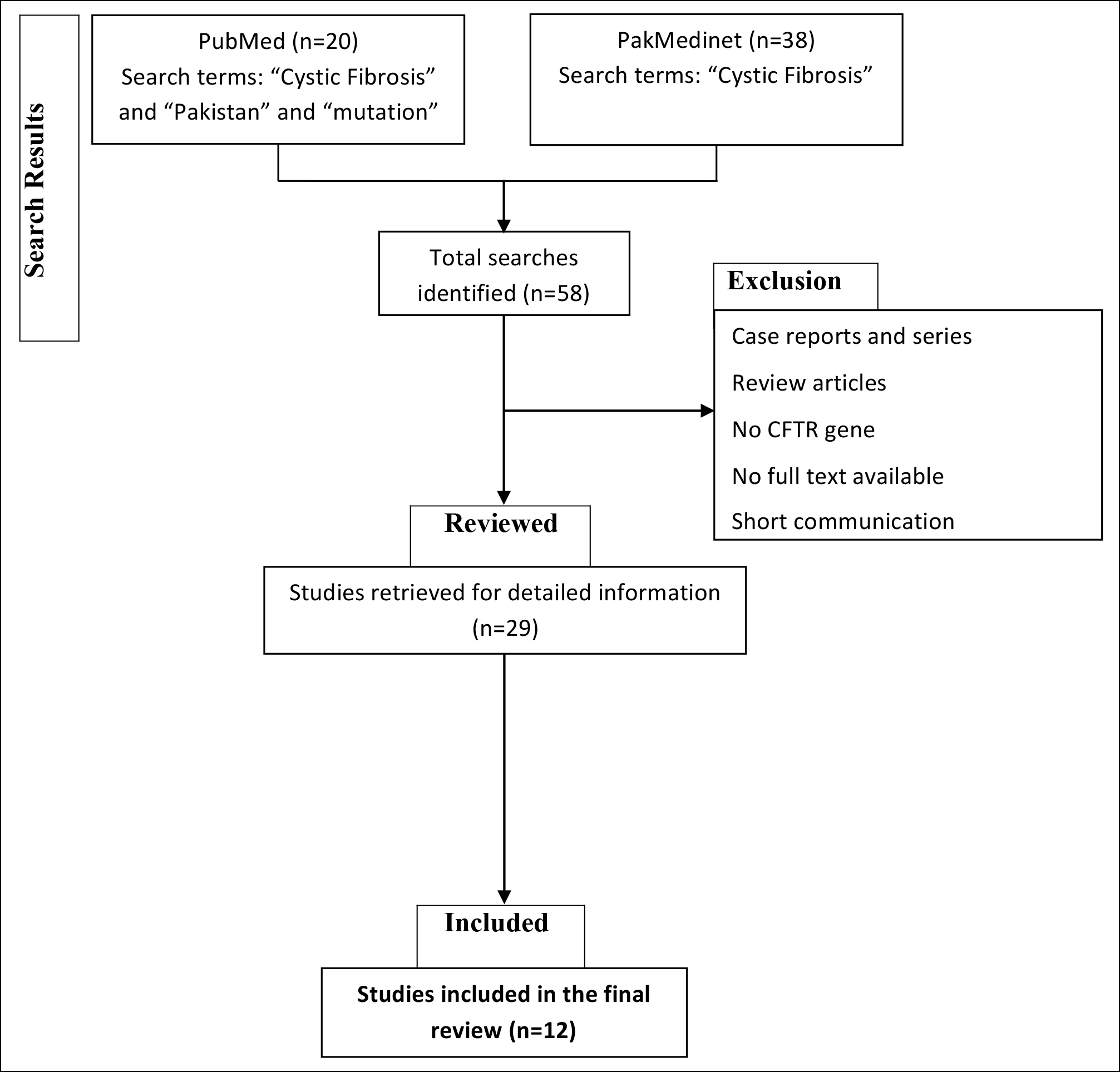 Figure 1: Consort diagram representing search, selection and inclusion of the studies (CF = Cystic fibrosis; CFTR = Cystic fibrosis transmembrane regulator).
Figure 1: Consort diagram representing search, selection and inclusion of the studies (CF = Cystic fibrosis; CFTR = Cystic fibrosis transmembrane regulator).
RESULTS
The search identified a total of 63 studies, PubMed (n= 18) and PakMedinet (n=45). Of them, twelve studies reporting CF gene mutations in Pakistani-origin patients were identified, findings are shown in Table I. The most widely tested mutation was Delta F508 with a reported frequency of 17-60%, but the spectrum of mutations identified was very wide. Thirty-three mutations were reported in exons 4-14, 18, and 19 and introns 2, 4, 6b, 10, and 19, shown in Table II.
DISCUSSION
This study reports the spectrum of CFTR gene mutations in Pakistani descent CF patients and shows evidence that Delta 508 may or may not be present in CF. The Delta 508, a disease causing mutation was present in 17-60% of the Pakistani CF patients only. The sweat chloride test is considered the gold standard for CF diagnosis but the results can be normal to indeterminate in some patients. In these patients’ DNA, mutational analysis is required for confirmation of CF. To confirm the diagnosis of CF, patients’ sample for CFTR gene mutations are sent to the other countries. There are two options available for these patients, either to get next-generation sequencing done for the complete CFTR gene (which is very expensive) or a cost-effective test to get tested for a common CFTR gene mutations panel including 23 mutations, recommended by the American College of Medical Genetics. The problem, however, with this panel is that it includes the common mutation reported in the Caucasian population, which may or may not be relevant to this population.
The literature reporting CFTR gene mutations is shown in Table I.11-21 Although D508 was the most commonly detected CFTR gene mutation, however, multiple other mutations were also reported in the Pakistani population (Table I). Many reported mutations are not associated with CF-related clinical signs and symptoms. The mutations reported in the Pakistani population on CFTR gene analysis and whether they are disease-causing mutations or not are shown in Table II.
A study done on 150 clinically suspected CF and with abnormal sweat chloride test patients in our population performed screening of DF508, S549R, S549N, Y569D, 296 + 12(T>C), G553X, G551D, and G551X mutations by allele specific-PCR and direct DNA sequencing of exons 10, 11, and 12 of CFTR gene. They reported that only 17% were positive for Delta 508 mutation while S549 was the second most common mutation.10 An observational study was conducted in a pediatric chest clinic in an urban tertiary care center in north India. In this study, clinical characteristics of the 120 children diagnosed with CF by quantitative pilocarpine iontophoresis were recorded. A polymerase chain reaction based test for identification of Delta F 508 mutation was performed on all children showed that only 19% of these patients had delta 508 mutation; considered one of the most common mutation in the Caucasian population.20
A study done in the UK reported that common genotypes in Indian subcontinent (ISC) origin patients were very different compared to the Caucasians.22 Delta F508 mutation was the most commonly reported mutation and 1 in 4 ISC patients had no CFTR gene mutation identified. This shows that there is a need to identify mutations common in this population and once identified, they can be grouped to form a panel. This panel can then be utilised for the diagnosis of CF in the Pakistani population. A panel consisting of common mutations reported in other populations will be ineffective to screen mutations in Pakistanis due to the genetic heterogeneity. There is a need to identify disease-causing mutations in Pakistani population by genotype-phenotype relationship studies.
Table I: Literature reporting CFTR gene (chromosome 7) mutations in Pakistani population.
|
|
Year |
Sample size (n) |
Number of Pakistani patients |
Molecular method of diagnosis |
CFTR gene mutation |
|
Bowler et al. 12 |
1993 |
27 |
9 Pakistani |
Genomic DNA analysis by PCR |
Delta F508 (present in 4/9 Pakistani) |
|
Curtis et al. 13 |
1993 |
666 |
1 Pakistani |
Genomic DNA analysis by PCR
|
G551D, R553X, and S549 Delta F508 |
|
Bhutta et al. 11 |
2000 |
15 |
15 Pakistani |
Delta F508 mutation analysis by Refractory Mutation System (ARMS) Amplification |
Delta F508 9/15 (60%) |
|
McCormick et al. 14 |
2002 |
5274 |
63 Pakistani |
Genomic DNA analysis
|
Delta F508: 35.3% 621+1G->T: 1.2% Y569D: 8.2% L218X: 3.5% 1161delC: 3.5% R709X/V456A: 2.4% G542X/G542X: 2.4 % |
|
Kabra et al. 20 |
2003 |
23 |
23 Pakistani-origin patients |
PCR based test for identification of Delta F508 mutation |
Delta F508 (56%) |
|
Mei-Zahav et al. 15 |
2005 |
381 |
2 Pakistani |
Screening of genomic DNA by allele specific oligonucleotide hybridisation. Extensive mutation analysis was then performed using PCR based multiplex heteroduplex analysis |
R117H/7T 3849+10kbC>T D1152H Delta F508: 13 L218X: 2 1525-1GRA: 1 S549N: 1 3849+10kbC-T: 1 V392G: 1 Unidentified 7 |
|
Shah et al. 15 |
2006 |
56 |
56 Pakistani |
Gene mutation analysis for Delta F508 by PCR |
Delta F508 (33%) |
|
Castellani et al. 17 |
2008 |
|
- |
Genomic DNA analysis
|
Y569D |
|
Shastri et al. 21 |
2008 |
16 |
16 Pakistani-origin patients |
Multiplex PCR |
Delta F508 (17 out of 33) |
|
Shah et al. 10 |
2009 |
150 |
150 Pakistani |
Mutations Delta F508, S549R, S549N, Y569D, 296 + 12(T>C), G553X, G551D and G551X were screened for by allele specific polymerase chain reactions. CFTR exons 10, 11 and 12 were sequenced by direct DNA sequencing. |
Delta F508: 26 S549R: 20 S549N: 1 Y569D 296 + 12(T>C) G553X G551D G551X |
|
Uppaluri et al. 18 |
2012 |
2 |
2 patients; 1 was of Pakistani descent |
Full sequencing of the CFTR gene |
V456A |
|
Aziz et al. 19 |
2017 |
43 |
43 patients |
Delta F508 mutation analysis by PCR |
Delta F508 |
Sweat chloride estimation by the coulometric method has a high incidence of false positive as well as false negative rates.23 Sweat sample collection is cumbersome and the test can also give indeterminate results.24 The chloride concentration in sweat is directly related to the sweat production rate so chances of false negative results increase at lower sweat production rates and the failure rate of sweat production is high in this population, 20-40%, while <5% is recommendation of regulatory bodies (unpublished data).25
There are certain challenges in diagnosing CF in Pakistan, mainly being that no diagnostic tests are available and diagnosis of CF is done based on clinical grounds. Very limited research is done in this area in Pakistan and none within the last five years. It is also not possible to estimate the burden of CF in the Pakistani population due to the non-availability of newborn screening and diagnosis is often delayed as well as the outcome of such patients is poor due to late diagnosis. Apart from this, lack of awareness among health care personals, no newborn screening services for CF, inequitable availability of medication, and specialty services are other limitations which lead to the poor outcomes of CF patients in the Pakistani population. There is a need to educate the masses to raise awareness of CF, establish newborn screening services for CF to determine the exact burden, increasing equitable access to CF diagnostic testing such as sweat chloride concentration analysis and CFTR gene mutations for early identification of CF patients, develop a population specific CFTR gene mutations panel for higher case detection, and develop specialty services for CF care for better patient’s outcome.
Table II: Mutations present in the CFTR gene (chromosome 7- q31.2), reported in Pakistani population on literature search.
|
Gene |
Exon / intron |
Protein name |
CDNA |
CF causing |
|
R117H15 |
Exon 4 |
p.Arg117His |
c.350G>A |
Varying clinical consequence |
|
L218X14,15 |
Exon 6 |
p.Leu218X |
c.653T>A |
Not reported in database |
|
1161delC14 |
Exon 8 |
p.Cys343X |
c.1029delC |
CF causing |
|
V392G15 |
Exon 9 |
p.Val392Gly |
c.1175T>G |
Not reported in database |
|
V456A14,18 |
Exon 10 |
p.Val456Ala |
c.1367T>C |
CF causing |
|
S466X (17) |
p.Ser466X |
c.1397C>A or c.1397C>G |
CF causing |
|
|
Delta50811-16,19-21 |
p.Phe508del |
c.1521_1523delCTT |
CF causing |
|
|
G542X14 |
Exon 11 |
p.Gly542X |
c.1624G>T |
CF causing |
|
G551D10,13 |
Exon 12 |
p.Gly551Asp |
c.1652G>A |
CF causing |
|
R553X13 |
p.Arg553X |
c.1657C>T |
CF causing |
|
|
S549R13 |
p.Ser549Arg |
c.1645A>C or c.1647T>G |
CF causing |
|
|
Y569D10,14 |
Exon 13 |
p.Tyr569Asp |
c.1705T>G |
CF causing |
|
R709X14 |
Exon 14 |
p.Arg709X |
c.2125C>T |
CF causing |
|
D1152H15 |
Exon 18 |
p.Asp1152His |
c.3454G>C |
Varying clinical consequence |
|
296 + 12(T>C)10 |
Intron 2 |
mRNA splicing defect |
- |
Not reported in database |
|
621+1G->T14 |
Intron 4 |
mRNA splicing defect |
c.489+1G>T |
CF causing |
|
1525-1G->A15 |
Intron 10 |
mRNA splicing defect |
c.1393-1G>A |
CF causing |
|
c.3849+10 kb C>T15 |
Intron 19 |
- |
c.3849+10 kb C>T |
Not reported in database |
CONCLUSION
The frequency of D508 mutations in the Pakistani population is low. The mutations reported in Pakistani population are different, the spectrum of mutations reported is wider. There is a need to identify common mutations in this population, perform genotypic-phenotypic correlations, and develop a population-specific mutations panel to improve the CF detection rate.
COMPETING INTEREST:
The authors declared no competing interest.
AUTHORS’ CONTRIBUTION:
HM: Conceived the idea, designed and conducted the project, and wrote the manuscript.
AHK: Supervised the project and critically reviewed the manuscript for intellectual content.
SBH: Did the literature search and wrote the manuscript.
TM: Critically reviewed the manuscript for intellectual content.
AN: Did the literature search and reviewed the manuscript.
All the authors have approved the final version of the manuscript to be published.
REFERENCES
- Farrell PM. The prevalence of cystic fibrosis in the European Union. J Cystic Fibrosis 2008; 7(5):450-3. doi.org/10.1016/j. jcf.2008.03.007.
- Kharrazi M, Yang J, Bishop T, Lessing S, Young S, Graham S, et al. Newborn screening for cystic fibrosis in California. Pediatrics 2015; 136(6):1062-72. doi.org/10.1542/peds.2015- 0811.
- Farrell PM, Rosenstein BJ, White TB, Accurso FJ, Castellani C, Cutting GR, et al. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic fibrosis foundation consensus report. J Pediatrics 2008; 153(2):4-14. doi. org/10.1016/j.jpeds.2008.05.005.
- Flume PA, Stenbit A. Making the diagnosis of cystic fibrosis. Am J Med Sci 2008; 335(1):51-4. doi.org/10.1097/MAJ.0b013e31815d2622.
- Goodchild M, Insley J, Rushton D, Gaze H. Cystic fibrosis in 3 Pakistani children. Arch Dis Child 1974; 49(9):739. doi.org/10.1136/adc.49.9.739.
- Prasad R, Sharma H, Kaur G. Molecular basis of cystic fibrosis disease: An Indian perspective. Indian J Clin Biochem 2010; 25(4):335-41. doi.org/10.1007/s12291- 010-0091-1.
- Hussain R, Bittles AH. The prevalence and demographic characteristics of consanguineous marriages in Pakistan. J Biosoc Sci 1998; 30(2):261-75. doi.org/10.1017/S00219 32098002612.
- Consortium CFGA. Cystic fibrosis mutation data base. http://www genet sickkids on ca/cftr/. 2003.
- Tsui LC, Dorfman R. The cystic fibrosis gene: A molecular genetic perspective. Cold Spring Harb Perspec Med 2013; 3(2):a009472. doi: 10.1101/cshperspect.a009472.
- Shah U, Frossard P, Moatter T. Cystic fibrosis: Defining a disease under‐diagnosed in Pakistan. Tropical Med Int Health 2009; 14(5):542-5. doi.org/10.1111/j.1365-3156. 2009.02253.x.
- Bhutta ZA, Moattar T, Shah U. Genetic analysis of cystic fibrosis in Pakistan: A preliminary report. J Pak Med Assoc 2000; 50(7):217.
- Bowler I, Estlin E, Littlewood J. Cystic fibrosis in Asians. Arch Dis Childhood 1993; 68(1):120-2. doi.org/10.1136/adc.68.1.120.
- Curtis A, Richardson R, Boohene J, Jackson A, Nelson R, Bhattacharya S. Absence of cystic fibrosis mutations in a large Asian population sample and occurrence of a homozygous S549N mutation in an inbred Pakistani family. J Med Gene 1993; 30(2):164-6. doi.org/10.1136/jmg.30.2.164.
- McCormick J, Green MW, Mehta G, Culross F, Mehta A. Demographics of the UK cystic fibrosis population: implications for neonatal screening. Eur J Hum Gene 2002; 10(10):583-90. doi.org/10.1038/sj.ejhg.5200850.
- Mei-Zahav M, Durie P, Zielenski J, Solomon M, Tullis E, Tsui L, et al. The prevalence and clinical characteristics of cystic fibrosis in South Asian Canadian immigrants. Arch Dis Childhood 2005; 90(7):675-9. doi.org/10.1136/adc.2003. 042614.
- Shah U, Moatter T, Bhutta Z. Profile and factors determining outcome in a cohort of cystic fibrosis patients seen at the Aga Khan University Hospital, Karachi, Pakistan. J Trop Pedia 2006; 52(2):132-5. doi.org/10.1093/tropej/fmi080.
- Castellani C, Cuppens H, Macek M, Cassiman J, Kerem E, Durie P, et al. Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. J Cystic Fibros 2008; 7(3):179-96. doi.org/10.1016/j.jcf.2008.03. 009.
- Uppaluri L, England S, Scanlin T. Clinical evidence that V456A is a cystic fibrosis causing mutation in South Asians. J Cystic Fibros 2012; 11(4):312-5. doi.org/10.1016/j.jcf. 2012.02.001.
- Aziz DA, Billoo AG, Qureshi A, Khalid M, Kirmani S. Clinical and laboratory profile of children with cystic fibrosis: Experience of a tertiary care center in Pakistan. Pak J Med Sci 2017; 33(3):554-9. doi.org/10.12669/pjms.333.12188.
- Kabra S, Kabra M, Lodha R, Shastri S, Ghosh M, Pandey R, et al. Clinical profile and frequency of delta f508 mutation in Indian children with cystic fibrosis. Indian Pediatric 2003; 40(7):612-9.
- Shastri SS, Kabra M, Kabra SK, Pandey RM, Menon PS. Characterisation of mutations and genotype-phenotype correlation in cystic fibrosis: Experience from India. J Cyst Fibros 2008; 7(2):110-5. doi.org/10.1016/j.jcf.2007.06.004.
- McCormick J, Ogston SA, Sims EJ, Mehta A. Asians with cystic fibrosis in the UK have worse disease outcomes than clinic matched white homozygous ΔF508 controls. J Cystic Fibros 2005; 4(1):53-8. doi.org/10.1016/j.jcf.2004.11.003.
- Denning CR, Huang NN, Cuasay L, Shwachman H, Tocci P, Warwick WJ, et al. Cooperative study comparing three methods of performing sweat tests to diagnose cystic fibrosis. Pedia 1980; 66(5):752-7. doi.org/10.1542/peds. 66.5.752.
- LeGrys VA. Sweat testing: Sample collection and quantitative analysis: Approved guideline: NCCLS 1994.
- Cirilli N, Padoan R, Raia V, Group ISTW. Audit of sweat testing: A first report from Italian cystic fibrosis centres. J Cystic Fibros 2008; 7(5):415-22. doi.org/10.1016/j.jcf. 2008.03.005/