Primary Renal Leiomyosarcoma
By Yunshen Ding1, Changyi Quan2Affiliations
doi: 10.29271/jcpsp.2021.06.725ABSTRACT
To optimise the diagnosis and treatment of primary renal leiomyosarcoma, we present our experience with a similar case and review the literature.
A 49-year woman was incidentally found to have a palpable mass in the right kidney. CT scan revealed an enhancing, heterogeneous, 6×7×9 cm right renal mass. On suspicion of renal carcinoma, a right radical nephrectomy was performed.
The pathological diagnosis was primary renal leiomyosarcoma. After 40 months of regular follow-up, local recurrence was found, and she had liver metastases. She died of cachexia due to tumor metastasis, and the survival period was just 44 months.
With unobvious clinical manifestations, preoperative imaging and postoperative pathological examination might be helpful for an accurate diagnosis. Radical nephrectomy to completely remove the tumor is recommended, and the combination of neoadjuvant or postoperative therapy should also be considered.
Key Words: Renal neoplasms, Leiomyosarcoma, Diagnosis, Therapy.
INTRODUCTION
Primary renal leiomyosarcoma is rare; and since it was reported by Berry in 1919, there have been several related reports in the domestic and foreign literature.1 Primary renal leiomyosarcoma is not only highly malignant but also demonstrates poor prognosis. Traditional examination cannot easily differentiate leiomyosarcoma from the sarcomatoid renal cell carcinoma. The incidence of this disease is low, and early clinical manifestations are not specific and obvious. Due to lack of characteristic imaging and molecular biology indicators, the prognosis of the relevant factors is still unclear. Hence, a diagnosis of primary renal leiomyosarcoma should be made with caution. We present our experience with a case of primary renal leiomyosarcoma managed at our hospital to further extend our understanding of the disease.
CASE REPORT
A 49-year woman was incidentally found to have a palpable mass in the right kidney with ipsilateral kidney tenderness by physical examination. She was admitted to our hospital in June 2014. There was no family history of cancer or chronic illness. A B-ultrasound showed a tumor with a cystic and solid hyperechoic area.
Moreover, blood vessels were detected within the tumor, as well as a lack of blood supply, which suggested a malignant tumor. CT scan showed an enhancing, heterogeneous, 6×7×9 cm right renal tumor with focal necrosis. The tumor density was uneven, slightly higher or lower than the renal parenchyma, and a CT value of 20-45 hounsefield units (HUs) was observed, with an average CT value of 40 HUs. Contrast-enhanced CT scanning showed varying degrees of enhancement, mild cortical enhancement, and a CT value of 32 - 65 HUs, with an average of 60 HUs (Figure 1). The delayed images demonstrated a significantly enhanced CT value of 50 - 72 HUs, with an average CT value of 68 HUs. Before operation, chest X-ray, ultrasound scan of the retroperitoneum and abdomen did not reveal signs of tumor dissemination. On suspicion of renal cell carcinoma, a right radical nephrectomy was performed. After surgery, she was treated with 2 courses of chemotherapy combined with "sandwich" type kidney area radiation (2 Gy per session) 20 times. Gross pathologic examination showed a mass measuring 6×7×9 cm, poorly demarcated from adjacent = renal parenchyma and involving the renal capsule. The cut surface showed gray white, solid and firm tumor mass, which demonstrated a cystic degeneration and areas of necrosis (Figure 2). Microscopically, the tumor was composed of spindle-shaped eosinophilic tumor cells arranged in parallel, interwoven and disorganised pattern (Figure 3). In addition, mitotic figures were frequent. Tumor cells showed infiltration of the kidney parenchyma and perirenal fat (Figure 4). Immunohistochemical staining demonstrated tumor cells to be positive for alpha-smooth muscle actin (α-SMA), desmin and vimentin. The pathological diagnosis was renal leiomyosarcoma. TNM staging showed pT2N0M0. Hence final diagnosis of primary renal leimyosarcoma was made. The postoperative course was uneventful. She presented with recurrence of tumor at 40 months after the radical operation and had liver metastases. She died of cachexia due to tumor metastasis, and the survival period was 44 months.
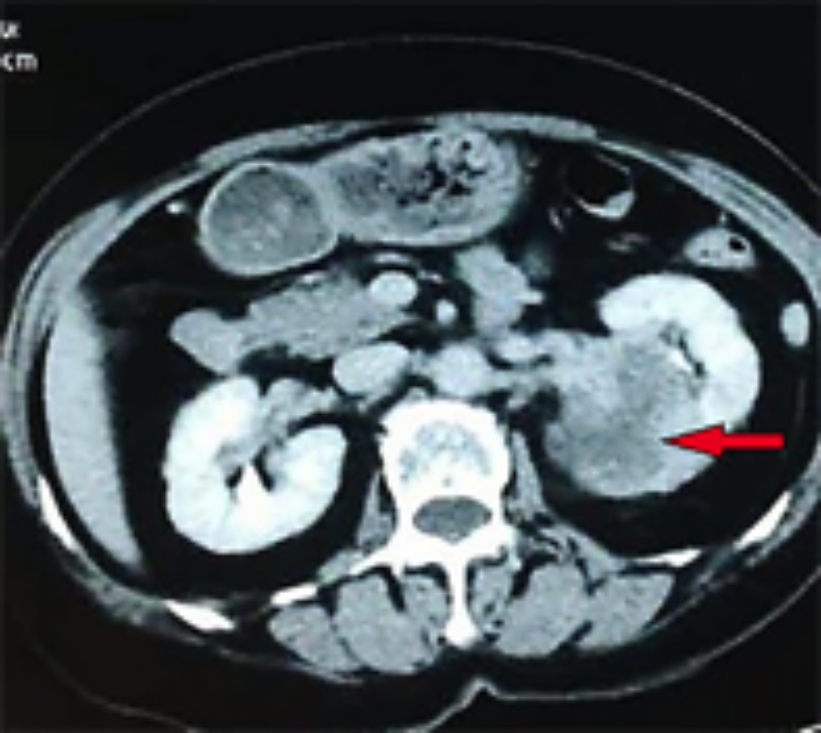 Figure 1: CT scan revealing an enhancing, heterogeneous, 6×7×9 cm, right renal mass (red arrow).
Figure 1: CT scan revealing an enhancing, heterogeneous, 6×7×9 cm, right renal mass (red arrow).
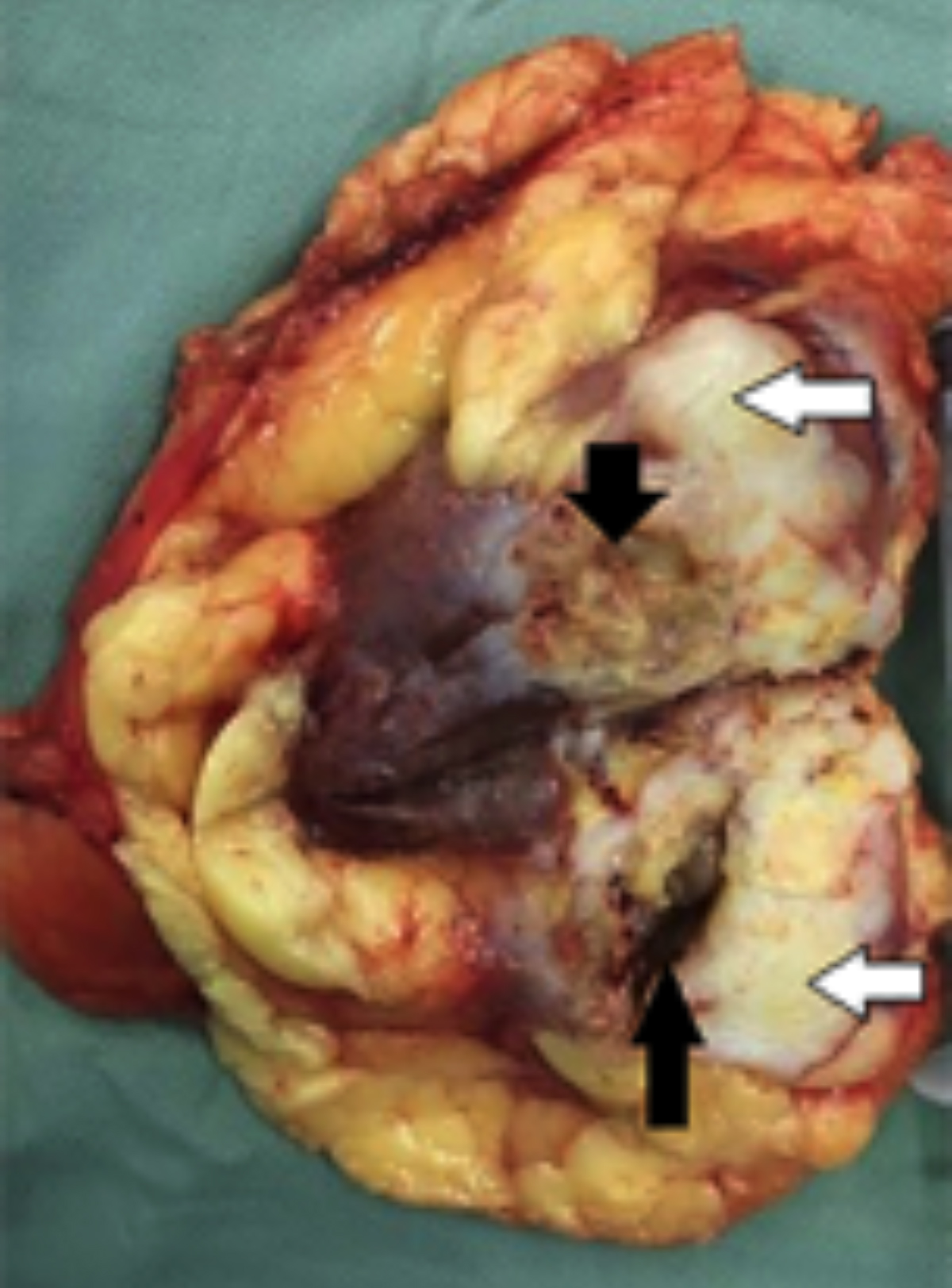 Figure 2: Cut section shows a gray-white, solid tumor (white arrow) with indistinct margins with surrounding renal parenchyma, and areas of necrosis and cystic degeneration (black arrow).
Figure 2: Cut section shows a gray-white, solid tumor (white arrow) with indistinct margins with surrounding renal parenchyma, and areas of necrosis and cystic degeneration (black arrow).
DISCUSSION
Adult primary renal sarcomas of rare tumor types, accounts for approximately 1 - 2% of all primary renal tumors. Of these, leiomyosarcoma is the most common histological subtype of renal sarcoma, accounts for 50% - 60% of later.2,3 These tumors can occur at any age and are more common in women aged 40 - 60 years.4 The incidence observed in females may be related to the deletion of one or more genes on the X chromosome, which evades X chromosome inactivation.5,6 By analysing 57 cases of renal leiomyoma and leiomyosarcoma characteristics, Gupta et al.7 observed that leiomyosarcomas exhibited a higher mitotic rate, Ki67-labelling index, and lack of expression of the estrogen receptor (ER) and progesterone receptor (PR). Primary renal leiomyosarcoma is genetically complex and usually shows a "chaotic" karyotype, including aneuploidy or polyploidy with no detectable recurrent tumor-specific translocations.8 In addition, cases have also been reported with the EBV virus infection, AIDS and after organ transplantation.4 In the present case, a 49-year woman, without obvious and specific clinical manifestations, was incidentally found to have a palpable mass in the right kidney by physical examination.
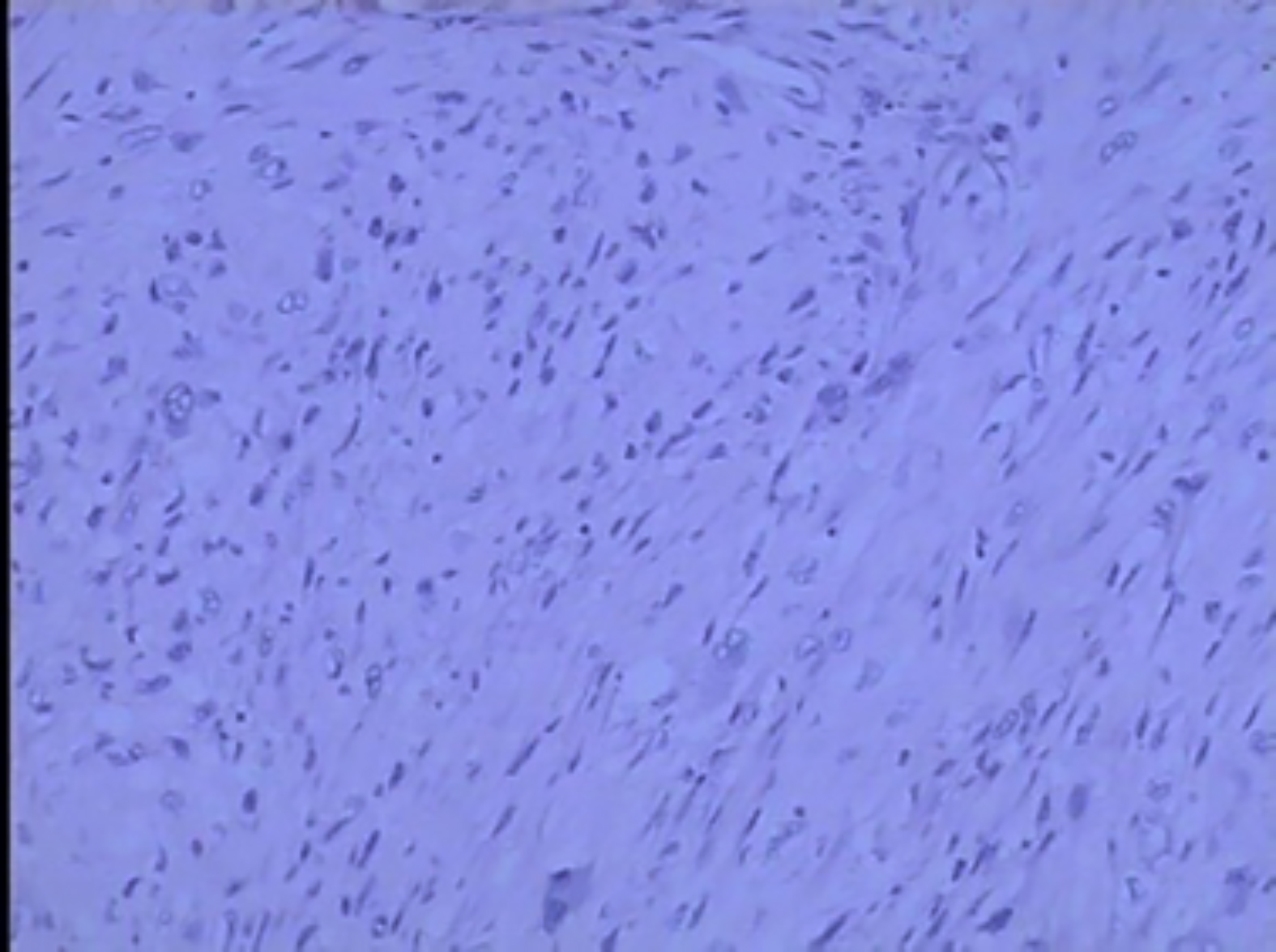 Figure 3: Low-power view showing spindle-shaped atypical eosinophilic tumor cells arranged in a parallel, interwoven or disorganized bundles (HE, ×100).
Figure 3: Low-power view showing spindle-shaped atypical eosinophilic tumor cells arranged in a parallel, interwoven or disorganized bundles (HE, ×100).
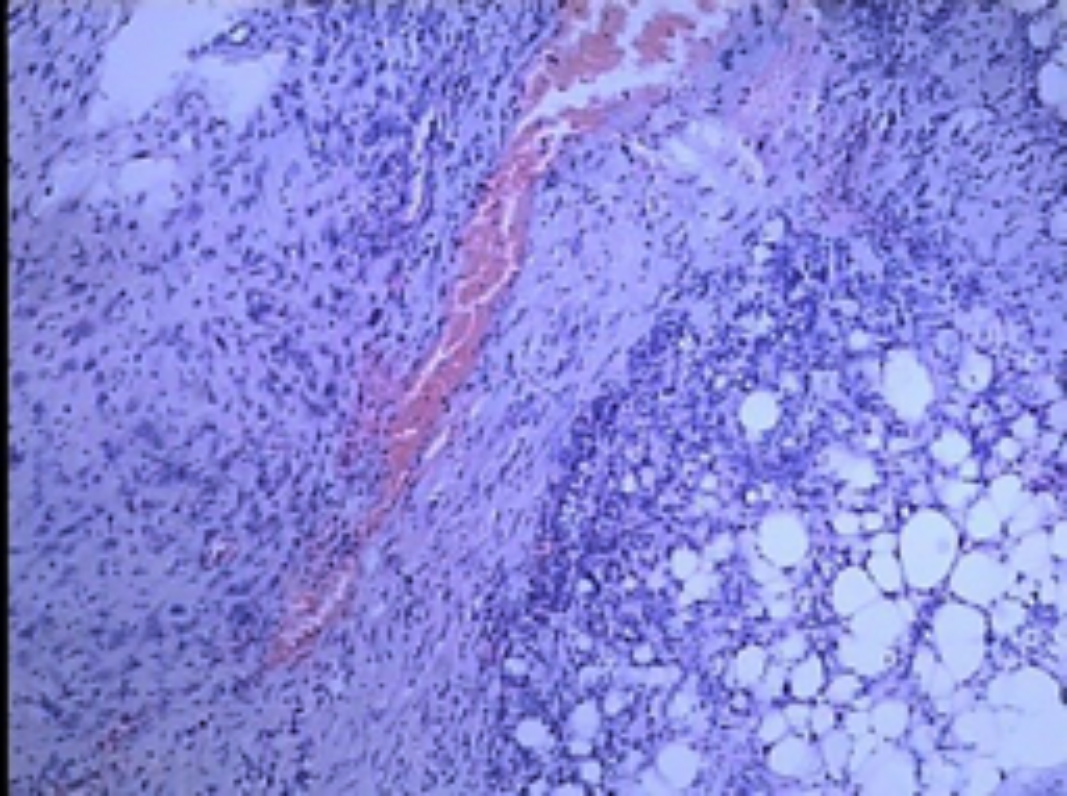 Figure 4: Tumor infiltration of renal and perirenal fat tissue (HE, ×100).
Figure 4: Tumor infiltration of renal and perirenal fat tissue (HE, ×100).
Renal leiomyosarcoma is most likely derived from the renal capsule, the renal pelvic smooth muscle cells, or blood vessels. Renal leiomyosarcoma has a high degree of invasiveness, high metastatic potential and often infiltrates renal parenchyma and renal sinus and perirenal adipose tissue.1,4,5,9 Microscopic examination reveals spindle cells, which are mainly arranged as intersecting bundles. Immunohistochemical staining was positive for α-SMA, desmin and vimentin; and tumor cells were invading the perirenal adipose tissue.
There were no significant clinical manifestations in this tumor. B-mode ultrasound showed a hyperechoic tumor, lacking blood supply. CT scan revealed an enhancing, heterogeneous, right renal mass. A provisional diagnosis of renal cell carcinoma was made. Due to rapid and aggressive growth and poor prognosis, primary renal leiomyosarcoma is more prone to metastasis in the early stage, and blood and lymph nodal metastases are common. Common metastatic sites are the liver and perirenal lymph nodes; however, other sites have been reported, such as lung, bone, stomach and soft tissue.1 In this case, the tumor stage of T2 was prone to metastasis.
Compared with other urinary tract sarcomas, renal sarcoma exhibits a poor prognosis. According to statistics, the 5-year survival rate of patients with retroperitoneal sarcoma was 82%, while the 5-year survival rates of patients with bladder sarcoma, prostate sarcoma, and renal sarcoma were 73%, 44%, and 39%, respectively.10 However, our patient did not survive for 5 years. Currently, radical nephrectomy is the primary treatment of primary renal leiomyosarcoma. Multivariate survival analysis was performed on 143 patients with primary retroperitoneal leiomyosarcomas by van Dalen et al.10
They concluded that complete tumor resection by radical surgery and low histology grade were the only factors affecting prognosis. However, there are also reports of partial nephrectomy with postoperative survival of 44 months.4 After 40 months of regular follow-up in our patient, local recurrence was found, and she had liver metastases. She died of cachexia due to tumor metastasis, and the survival period was 44 months. Adjuvant radiotherapy and chemotherapy may be used in the case of patients with renal leiomyosarcoma. Reports have indicated that chemotherapy (specifically gemcitabine and docetaxel) and radiotherapy may benefit patients' survival after radical nephrectomy or partial nephrectomy.4 In addition, phase II studies report the possibility of using a tyrosine kinase inhibitor, such as sunitinib for the treatment of primary renal leiomyosarcoma. Reports have indicated that combined treatment with chemotherapy or radiotherapy after radical surgery can not only prolong the tumor recurrence time but also slightly prolong the survival time. There is a need for more studies to strengthen the evidence base.
In summary, primary renal leiomyosarcoma is a rare, aggressive tumor of kidney with a poor prognosis. A high level of suspicion should be kept in mind when preoperative B-ultrasound shows a hyperechoic solid tumor with a lack of blood supply; whereas, the CT scan shows uneven density of tumor with delayed enhancement pattern. Pathology examination including immunohistochemistry is crucial in making correct diagnosis. For patients with tumor stage ≥ T2N0M0, radical resection should be performed as soon as possible; and regularly reviewed to detect and control tumor metastasis. Although complete surgical excision is the primary treatment for renal leiomyosarcoma, postoperative adjuvant therapy should also be considered to prolong the patient's survival.
ACKNOWLEDGMENTS:
We thank the members of our Department for their critical reading. This work was supported by National Nature Young Scientists Found of China 81502217, Tianjin Municipality: Plan of Funding Outstanding Science and Technology Projects launched by Talents Returning from Studying Overseas (20160014).
CONFLICT OF INTEREST:
The authors declared no conflict of interest.
AUTHORS’ CONTRIBUTION:
YD: Drafted the work or revising it critically for important intellectual content.
CQ: Agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
REFERENCES
- Narula V, Siraj F, Bansal A. Renal leiomyosarcoma with soft tissue metastasis: An unusual presentation.Can Urol Assoc J 2015; 9(3-4):E139-41. doi: 10.5489/cuaj.2396.
- Venkatesh K, Lamba Saini M, Niveditha SR, Krishnagiri C, Babu S. Primary leiomyosarcoma of the kidney. Patholog Res Int 2010; 2010:652398. doi: 10.4061/2010/652398.
- Tajima S, Waki M, Fukuyama M. Pleomorphic leiomyosarcoma with a dedifferentiation-like appearance in the kidney: Case report and literature review. Med Mol Morphol 2016; 49(4):238-42. doi: 10.1007/s00795-015-0103-6.
- Ozturk H. High-grade primary renal leiomyosarcoma. Int Braz J Urol 2015; 41(2):304-11. doi: 10.1590/S1677-5538.IBJU.2015.02.17.
- Tanaka T, Koie T, Iwabuchi I, Ogasawara M, Kawaguchi T, Ohyama C. Primary leiomyosarcoma of a horseshoe kidney in a woman with Turner syndrome: A case report. BMC Research Notes 2014; 7:491. doi: 10.1186/1756-0500-7-491.
- Evans D, Fowlkes N. Renal leiomyosarcoma in a cat. J Vet Diagn Invest 2016; 28(3):315-8. doi: 10.1177/1040638716638126.
- Gupta S, Jimenez RE, Folpe AL, Cheville JC. Renal leiomyoma and leiomyosarcoma: A study of 57 cases. Am J Surg Pathol 2016; 40(11):1557-63. doi: 10.1097/PAS.0000000000000681.
- Miettinen M. Smooth muscle tumors of soft tissue and non-uterine viscera: Biology and prognosis. Mod Pathol 2014; 27(Suppl1): S17-29. doi: 10.1038/modpathol.2013.178.
- Nicolas MM, Tamboli P, Gomez JA, Czerniak BA. Pleomorphic and dedifferentiated leiomyosarcoma: Clinicopathologic and immuno-histochemical study of 41 cases. Hum Pathol 2010; 41(5):663-71.
- van Dalen T, Plooij JM, van Coevorden F, van Geel AN, Hoekstra HJ, Albus-Lutter C, et al. Long-term prognosis of primary retroperitoneal soft tissue sarcoma. Eur J Surg Oncol 2007; 33(2):234-8. doi: 10.1016/j.ejso.2006.09.020.