Myoclonic Astatic Resistant Epilepsy and Disproportionate Overgrowth Carrying a Duplication in 2q13 and Deletion in the 6p21.32 Chromosomal Regions
By Sevim Turay1, Recep Eroz2, Ebru Karagun3Affiliations
doi: 10.29271/jcpsp.2022.06.808ABSTRACT
Copy number variants have been increasing due to a rise in the availability of array comparative genomic hybridisation, which occupies an important place in diagnosis, especially in patients with epilepsy, dysmorphic findings, and intellectual disability. We detected 2q13 chromosomal duplication and 6p21.32 chromosomal deletion in a patient under follow-up due to epilepsy, developmental retardation, dysmorphic findings, and asymmetric overgrowth in our clinic since the age of six months. The parents had only 2q13 mutations. Copy number variation in 2q13 is associated with dysmorphic findings, psychiatric disorders, and developmental delays. However, the exact pathogenicity is not yet known. We think that 6p21.32 chromosomal deletion caused resistant epilepsy and lipodystrophy in this patient. We anticipate that this case will contribute to the literature by linking disorders caused by the current chromosomal abnormality to clinical findings.
Key Words: Myoclonic astatic seizure, Resistant epilepsy, Dermal atrophy, Chromosomal microarray analysis.
INTRODUCTION
Copy number variations (CNVs), whose pathogenicity is not well understood, are continuing to rise with the frequent use of the array comparative genomic hybridization (aCGH) technique. Numerous studies in recent years have shown the sensitivity of aCGH in patients with intellectual disability and dysmorphic findings, and have recommended its use as a first step screening test.1 The importance of aCGH in the diagnosis of epilepsy is also well known.2 This study describes a non-consanguineous Turkish family consisting of four members. Peripheral blood of the patient was obtained after informed consent was received from the parents for chromosomal analysis. Since the patient had a normal karyotype (46,XY), genomic DNA was isolated from peripheral blood samples and Chromosomal Microarray Analysis (CMA) was performed with Agilent ISCA v2 Human Genome 8x60k oligonucleotide array. A gain of 0.3 MB in 2q13 (110862477-111128847) ×3 and a 0.2 MB loss in 6p21.32 (32688930-32956557) ×1 were detected (Figure 1).
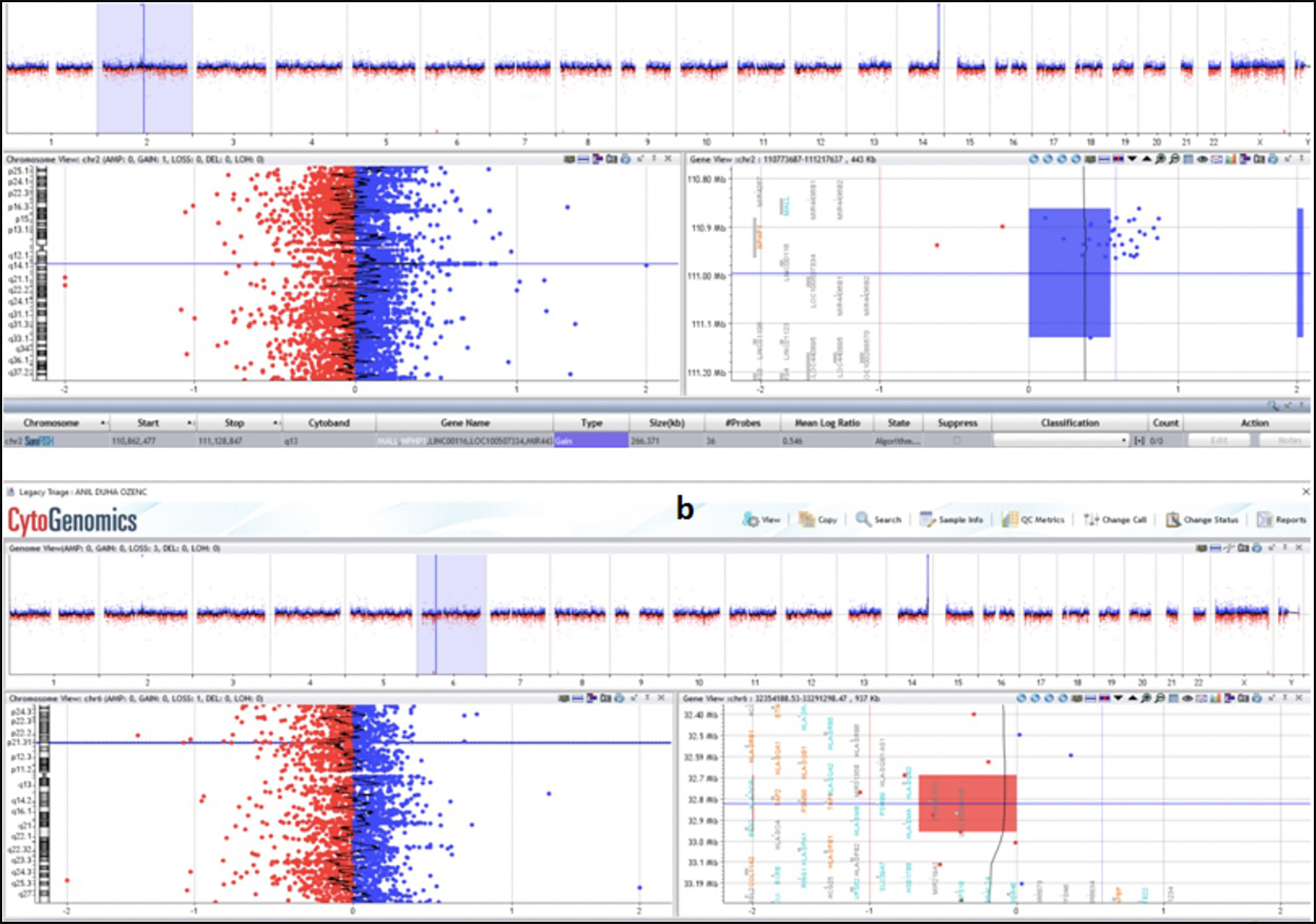 Figure 1: Chromosomal microarray analysis (CMA) results of the case performed by agilent ISCA v2 human genome 8x60k oligonucleotide array. A gain of 0.3 MB in 2q13 (110862477-111128847) ×3 (a), 0.2 MB loss in 6p21.32 (32688930-32956557) ×1 (b).
Figure 1: Chromosomal microarray analysis (CMA) results of the case performed by agilent ISCA v2 human genome 8x60k oligonucleotide array. A gain of 0.3 MB in 2q13 (110862477-111128847) ×3 (a), 0.2 MB loss in 6p21.32 (32688930-32956557) ×1 (b).
CNV in 2q13 was detected based on dysmorphic findings, psychiatric disorders, and developmental delays.3 However, the exact pathogenicity is not yet known, and the present finding was defined as a variant of uncertain significance. This report describes a case with chromosomal micro-deletion and duplication, together with a discussion of the affected gene and clinical findings in the light of the current literature.
CASE REPORT
A seven-year boy had been under follow-up due to treatment-resistant epilepsy with myoclonic astatic and perioral/eye myoclonus and had been using multiple anticonvulsants since the age of six months. Physical examination revealed an asthenic appearance (BMI 13.4 kg/m2), a flattened nasal root, asymmetric, disproportionate overgrowth (macrodactyly in the second and third left toes), and soft tissue swelling extending from the toes to the soles of the feet (Figure 2). The social communication was adequate, and speech, hearing, and vision examinations were normal. The muscle strength was good, and no limited mobility, hepatosplenomegaly, murmur or skin lesions were observed. His performance at school was poor and was therefore receiving special education.
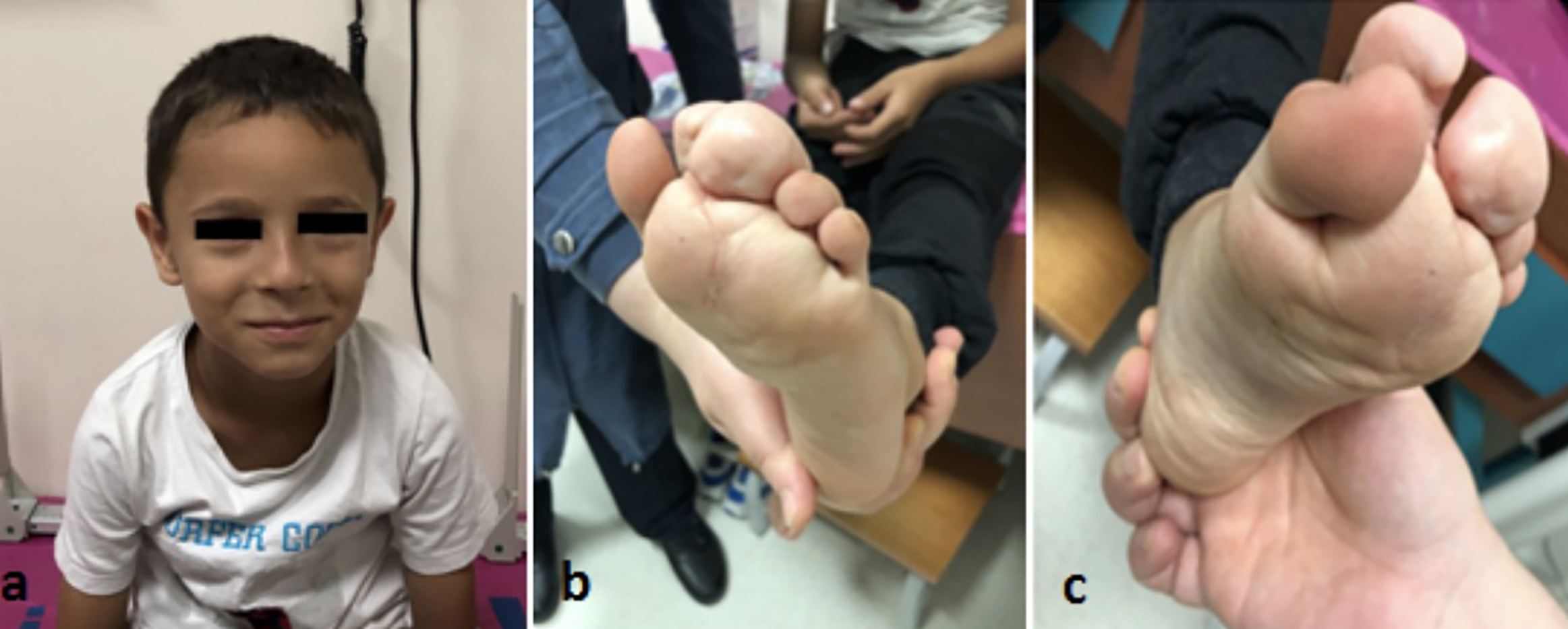 Figure 2: Flattened nasal root (a), macrodactyly in left foot 2nd and 3rd fingers (b,c), soft tissue swelling extending from the soles of the toes to the soles of the feet (b,c).
Figure 2: Flattened nasal root (a), macrodactyly in left foot 2nd and 3rd fingers (b,c), soft tissue swelling extending from the soles of the toes to the soles of the feet (b,c).
The patient was born at 39 weeks’ gestation with a birth weight of 3000 g by normal spontaneous vaginal delivery to a 31-year mother. At the age of one year, he was operated due to syndactyly in the second and third left toes. No history of hypoxia, frequent infection, or recurrent fever was present. The patient had a healthy 14-year brother. No illness or epilepsy was present in other family members. Complete blood count, biochemical parameters and abdominal ultrasound, and echocardiography were normal. Repeated encephalograms (EEGs) were compatible with myoclonic epilepsy. Cranial magnetic resonance imaging was normal. Biopsy taken from the tissue in the sole of the foot revealed dermal atrophy compatible with fat lobules in the mid-dermis and increased collagen tissue. Based on the histopathological findings, the patient was diagnosed with connective tissue nevus. Proteus syndrome was therefore suspected, and DNA was initially obtained from the patient’s peripheral blood sample. No mutation was detected after the whole exome screening of the akt1 gene. DNA was therefore re-obtained from the biopsy material since the cells with mutation in the toes with hyperplasia only would continue to grow and divide, while this would not occur in the other cells (mosaicism), and the akt1 gene was sequenced. No mutation was again detected at the whole exome screening of the akt1 gene. The patient had a normal karyotype (46, XY). CMA analysis revealed a gain of 0.3 MB in 2q13 (110862477 - 111128847) ×3 and a 0.2 MB loss in 6p21.32 (32688930 - 32956557) ×1 was detected (Figure 1). The parents exhibited a gain of 0.3 MB in 2q13 (110862477 - 111128847), but no loss in 6p21.32 (32688930 - 32956557) ×1.
DISCUSSION
The Mall and nphp1 genes described in OMIM are located in the 6p21.32 (32688930 - 32956557) region. The hladqa2, hladqb2, hladqb, tap2, psmb8, tap1, psmb9, hla-dmb, hla-dma and brd2 genes described in OMIM are located in the 2q13 (110862477 - 111128847) region. The parents of the present patient exhibited a gain of 0.3 MB in 2q13 (110862477 - 111128847) region. A study from 2018 reported 21 deletions and four duplications in the chromosome 2q13 region found to be associated with mild mental insufficiency and childhood psychiatric diseases.3 This patient had attention deficit and a dull intelligence level according to the WISC-R scale. The parents’ intelligence levels were normal. That study also examined previously detected chromosomal deletions (54) and duplications (23), and then grouped the accompanying findings.
Duplication was found not to be accompanied by heart abnormalities and convulsions, but rates of dysmorphic findings, intellectual disability, and developmental delay were high.3 This patient's findings were accompanied by epilepsy, but with no facial appearance dysmorphia. Duplication accompanied by autistic spectrum disorder, intellectual disability, and liver disorder has also been reported in two brothers. However, the responsible genes were reported as mall, nphp1, rgpd6, and bub1, although the same copy number change was smaller in the father, and the only affected gene was nphp1.4 The same regions (mall and nphp1) were also affected in our patient and the parents. In OMIM, the nphp 1 gene has been associated with Joubert syndrome-4, Nephronophthisis-1 juvenile, and Senior-Loken syndrome. However, no findings suggestive of these diseases were observed in the present case.
This review of the literature revealed similar information concerning the CNV located at 6p21.32. However, the psmb8 gene, one of the genes related to this region, has been identified as autosomal recessive in proteasome-associated auto inflammatory syndrome-1 (PRAAS1) in OMIM, a several studies on the subject have been published. This disease is accompanied by high fever, atypical neutrophilic dermatosis, lipodystrophies, and joint contractures caused by immune dysregulation, and is also known as chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE).5 Interestingly, Liu et al. reported the same clinical findings in two patients with heterozygous mutation. No additional gene pathology capable of causing this condition was detected at advanced genetic testing.6 Brehm et al. reported two cases with double heterozygous mutations.5 We speculated that the lipodystrophy in our patient might be caused by psmb8. Heterozygous formation may explain the absence of other symptoms of candle syndrome. In OMIM, the 6p21 region has been described together with juvenile myoclonic epilepsy (JME) 3, and a predisposition to JME has been reported in defects in this region. A large JME-sensitive locus has been associated with the chromosome 6p21 region.7 Pal et al. also suggested that the brd2 gene was the main susceptibility gene for JME.8 We also thought that this might account for the resistant myoclonic epilepsy in our patient. We were unable to establish a relationship between the other genes in 6p21 and our patient's clinical signs. Unfortunately, further investigation was not possible due to financial and technical constraints.
In conclusion, 2q13 duplication and 6p21.32 deletion were detected in a patient under follow-up due to resistant myoclonic epilepsy, developmental retardation, and dysmorphic findings. We think that the data from this case will make a significant contribution to the existing literature.
ACKNOWLEDGMENTS:
This study was completed in collaboration between the Duzce University Medical Faculty Paediatric and Genetic Medicine Departments, Turkey. We are most grateful to all the staff members and the patient’s family for their assistance with this report.
PATIENT'S CONSENT:
Consent to the publication of the patient’s clinical data and photographs was granted by both parents. Genetic analysis was performed after written informed consent from both parents.
COMPETING INTEREST:
The authors declared no competing interest.
AUTHORS’ CONTRIBUTION:
ST, RE: Original concept of study and critical review of manuscript.
ST, RE, EK: Collection, recording, analysis, and interpretation of data, and writing of manuscript.
All authors approved the final version of the manuscript to be published.
REFERENCES
- Miller DT, Adam MP, Aradhya S, Biesecker GL, Brothman AR, Carter NP, et al. Consensus statement: Chromosomal microarray is a first- tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 2010; 86(5):749-64. doi: 10.1016/j.ajhg.2010.04.006.
- Olson H, Shen Y, Avallone J, Sheidley BR, Pinsky R, Bergin AM, et al. Copy number variation plays an important role in clinical epilepsy. Ann Neurol 2014; 75(6):943-58. doi: 10.1002/ana.24178.
- Wolfe K, McQuillin A, Alesi V, Boudry Labis E, Cutajar P, Dallapiccola B, et al. Delineating the psychiatric and behavioral phenotype of recurrent 2q13 deletions and duplications. J Med Genet B Neuropsychiatr Genet 2018; 177(4):397-405. doi: 10.1002/ajmg.b.32627.
- Chen CP, Lin SP, Lee CL, Chern SR, Wu PS, Chen YN, et al. Recurrent 2q13 microduplication encompassing MALL, NPHP1, RGPD6, and BUB1 associated with autism spectrum disorder, intellectual disability, and liver disorder. Taiwan J Obstet Gynecol 2017; 56(1):98-101. doi: 10.1016/j.tjog. 2016.12.003.
- Brehm A, Liu Y, Sheikh A, Marrero B, Omoyinmi E, Zhou Q, et al. Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J Clin Invest 2015; 125(11):4196-211. doi: 10.1172/JCI81260.
- Liu Y, Ramot Y, Torrelo A, Paller AS, Si N, Babay S, et al. Mutations in proteasome subunit beta type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum 2012; 64(3): 895-907. doi: 10.1002/art.33368.
- Sander T, Bockenkamp B, Hildmann T, Blasczyk R, Kretz R, Wienker TF, et al. Refined mapping of the epilepsy susceptibility locus EJM1 on chromosome 6. Neurol 1997; 49(3):842-47. doi: 10.1212/wnl.49.3.842.
- Pal DK, Evgrafov OV, Tabares P, Zhang F, Durner M, Greenberg DA. (BRD2 (RING3) is a probable major susceptibility gene for common juvenile myoclonic epilepsy. Am J Hum Genet 2003; 73(2):261-70. doi: 10.1086/ 377006.