Highly Sensitive and Ultrafast Liquid Chromatography-tandem Mass Spectrometry Internal Standard Method for Determination of Alpha-Tocopherol in Serum and Application of Validated Method on Patient Serum Samples in Pakistan
By Humera Shafi Makhdoom, Mohammad Dilawar Khan, Omar Rasheed Chughtai, Zeerak Abbas, Areeba Azad, Hijab BatoolAffiliations
doi: 10.29271/jcpsp.2023.03.254ABSTRACT
Objective: To optimize and validate a specific, sensitive and fast liquid Chromatography coupled to triple quadrupole Mass Spectrometric (LC-MS / MS) technique for accurate detection of serum α-tocopherol (Vitamin E) levels.
Study Design: An experimental based study.
Place and Duration of Study: The Clinical and Forensic Toxicology section of Chughtai Lab, Jail Road Lahore, from April to September 2022.
Methodology: Methanol was used to deproteinize serum samples. The chromatographic separation was achieved using an Agilent Infinity-Lab Poroshell 120EC-C18 column, Agilent 6470 LC-MS/MS (equipped with an Electron Spray Ionization source) in gradient elution mode using 0.1% LCMS grade formic acid in water and LCMS-grade methanol as mobile phases. Hexa-deuterated α-tocopherol was employed as internal standard to minimise matrix interferences.
Results: The retention time of α-tocopherol was 3.0 ± 0.1 minutes. The linear concentrations obtained were ranged from 0.05-2 mg/dL with ≥0.985% coefficient of linearity. Detection and lower quantification limits determined were 0.025mg/dL and 0.05mg/dL, respectively. Recovery ranged from 96.5 to 99.8% and ionization suppression was -15.2% and -15.9% at high and low concentrations of α-tocopherol in serum. Intra-day and inter-day coefficient variation values were 4.2-4.9% and 5.0-5.9%, respectively.
Conclusion: An efficient and reliable tandem mass spectrometric technique for vitamin E analysis in serum was optimized, validated, and applied to 80 patient samples. This method has usefulness in clinical application for the accurate determination of vitamin E without potential matrix interferences.
Key Words: Vitamin E, LC-MS/MS, Tocopherol, Internal standard, Validation.
INTRODUCTION
Vitamin E, a micronutrient that belongs to fat-soluble vitamins has a crucial significance in the realisation as well as regulation of physiological activities in human body. Naturally, four forms of vitamin E (α-, β-, γ-, and δ-tocopherols) and tocotrienols (unsaturated-analogues of the four forms of tocopherol) exists. The α-Tocopherol is the most pharmacologically active form and suitable biomarker for in-vivo evaluation of vitamin E status. It acts as a peroxyl radical scavenger and has protective role within membranes and lipoproteins.1
The antioxidant function of α-Tocopherol has significance in the prevention of oxidative stress-associated diseases such as diabetes mellitus, cancer, cataracts, cardiovascular, central neuro-degenerative, and age-associated macular degeneration diseases.1,2 The protective role of fat-soluble vitamins in some cancers and coronary heart diseases has been suggested by epidemiological studies whereas few studies have indicated increased mortality linked to excessive vitamin E use but it has disputed acceptance.3-6
Accurate and precise measurement of vitamin E in clinical laboratories is quite difficult due to the molecular structural properties of the vitamin, availability of certified reference material, standard measurement quantification methods, and reference testing laboratories. The blood concentration of vitamin E is relatively variable and low due to the very small size of the molecule and the unavailability of specific information on vitamin stability in blood.7,8 Existing studies regarding the stability of vitamin E in blood provide contradiction in findings. For instance, one study claimed that the change in blood tocopherol level at ambient temperature was -1.0% for 72 hours while another study reported it as 4.8%.9,10 Therefore, even after using the same method the variation in results of different laboratories exists.
Different extraction and analysis methods for serum vitamin E determination have been reported historically.11-19 These methods involved laborious extraction procedures and high solvent consumption which rendered them time-consuming and expensive to use in clinical laboratories. More commonly utilised methods for estimation of serum vitamin E are reversed phase liquid chromatography methods coupled to ultraviolet-visible or diode array detectors.11,12 The major limitation is inability in separation of co-eluted interferences with liquid chromatography which can be resolved by the use of mass spectrometry. Vitamin E is a hydrophobic molecule and carrier proteins in blood help in its transportation. An accurate and precise estimation of this vitamin requires the breakage of vitamin-protein bond linkage. Further, it is a too small molecule (<500Da) having a single hydroxyl group that renders its ionization difficult.
In mass spectral analysis, ionization is one of the most critical steps.13 Liquid chromatography equipped with a triple quadrupole mass spectrometer (LC-MS/MS) is a sensitive technique as compared to HPLC methods.14,15 This technique combines the resolving power of high-pressure liquid chromatography with highly selective detection of mass spectrometer. This combination allows more precise and accurate quantification of vitamin E. This fast analytical method has additional benefits of reduced specimen volume and flow rate of mobile phases which further accounts for its cost-effectiveness. The objective of this study was to optimise and validate the clinical UHPLC-MS/MS technique for the determination of serum α-tocopherol levels.
METHODOLOGY
Certified reference materials of α-tocopherol (1 mg/mL) and α-tocopherol d6 (500 µg/mL) were purchased from Cerilliant, Round Rock, Texas, USA. LCMS grade methanol, acetonitrile, water, and formic acid were bought from Merck Chemicals. Analytical grade monobasic sodium phosphate and n-hexane were purchased from Merck Chemicals. SeraCon-DL matrix from SeraCare (Massachusetts, USA) was used as blank matrix for vitamin E. The study was started in April 2022 and completed in September 2022. An Agilent 1260 Infinity II Ultra High Pressure Liquid Chromatograph (UHPLC) equipped with 6470 tandem mass spectrometer (MS/MS) was employed for the estimation of vitamin E in the serum samples. Reversed phase C-18 analytical column (Agilent Infinity-Lab Poroshell 120EC-C18; 2.1mm x 50mm x 2.7µm) was utilised to achieve chromatographic separation. LCMS grade methanol and 0.1% formic acid in water were used as mobile phases after filtration through a 0.2µm nylon filter and degassing in an ultrasonic bath. For the elimination of phospholipids co-eluted with vitamin E and to achieve peaks with high resolution, gradient elution mode was employed. Separation was carried out at the column temperature set at 35oC with mobile phase flow rate of 0.3mL/min. Other instrumental operating parameters are presented in Table I.
Stock solutions of α-tocopherol (10mg/dL) and α-tocopherol d6 internal standard (50 µg/mL) in methanol were prepared and stored at -20°C. Stock solution of α-tocopherol and α-tocopherol d6 were diluted with methanol to prepare working standard solution of α-tocopherol (2.5 mg/dL) and working internal standard solution (5µg/mL) of α-tocopherol d6. Working control solution of α-tocopherol (2.5 mg/dL) was prepared by dilution of the stock solution of α-tocopherol with LCMS grade methanol. These solutions could be stored for six months without significant loss of vitamin E at -20°C and for three months at 4°C, if protected from light. Five calibration levels (0.05, 0.25, 0.5, 1.0, and 2.0 mg/dL) and two positive control samples (0.75 and 1.5 mg/dL) of α-tocopherol were prepared by spiking working standard solution of α-tocopherol (2.5 mg/dL) in SeraCon-DL matrix. SeraCon-DL matrix was also used as negative quality control sample. The calibration levels, positive and negative controls were extracted using the following procedure:
To 100 µL serum sample in an eppendorf tube, 50 µL 0.1M Phosphate buffer (pH 7.2), and 100 µL methanol were added. The tube was vortexed for 30 seconds and then centrifuged at 13000 rpm for 15 minutes. Two hundred µL of supernatant from eppendorf tube was transferred to another labelled eppendorf tube, and 25 µL working internal standard solution and 1000 µL n-hexane were added. The tube was capped tightly, vortexed for 30 seconds, rotated for 10 minutes to ensure maximum partitioning of analyte in two immiscible liquid layers and then centrifuged at 4000 rpm for 15 minutes. Hexane layer was transferred to a glass culture tube and subjected to complete evaporation using nitrogen evaporator at 50°C. After evaporation, it was reconstituted with 100µL methanol (LCMS grade) and transferred to an amber-colored LC-vial containing 150 µL glass vial insert. The LC vial was screw capped and analysed on LC-MS/MS using operating parameters mentioned in Table I.
The proposed method had been validated according to the Clinical and Laboratory Standards Institute document C62-A, FDA-guidelines on bio-analytical method validation, and Scientific Working Group Toxicology validation guidelines. Calibration curves were plotted using five calibration levels of α-tocopherol in SeraCon-DL matrix. Each level was run in triplicate for five days. The ratio of α-tocopherol peak area to its internal standard vs. the nominal spiked concentrations by a linear least squares regression model was used. The coefficient of determination (r2) was ≥0.985. All the calibration levels must have ≤20% precision (% CV) and ±20% accuracy for α-tocopherol. The lowest precise and accurate concentration of the calibration curve was accepted as lower limit of quantitation (LLOQ). LLOQ precision and accuracy were kept at ≤30% and ±30%, respectively. The limit of detection (LOD) was evaluated from a linear calibration curve or designated as half of the lowest calibration level for practical reasons. LOD concentration was analysed in triplicate for five days in order to determine precision. LOD precision must be ≤20%. To validate selectivity, high levels of ϒ-tocopherol (50 µg/ml, analog for α-tocopherol) were spiked into low-QC and high-QC samples to fabricate interference-introduced serum.
After analysis, the recovery of the α-tocopherol in these positive control samples should remain at ±20% of target concentration. Both accuracy and precision were investigated at low, medium, and high concentration levels (0.15, 0.75 and 1.5mg/dL of α-tocopherol) in serum sample. Each level was run in triplicate for five days to determine the inter-day and intraday precision. The values of accuracy were kept at ±20% of target concentration and precision at ≤20%. Pre- and post-injections of back-ground matrix with the highest calibration level were studied to evaluate the carry-over effect. The results were targeted at less than 15% of LLOQ. The ion suppression / enhancement study or matrix interference was evaluated by comparing the results of two different sets of samples. The α-tocopherol and internal standard peak areas of neat standard samples were compared to matrix samples fortified with neat standards after extraction. Both groups were spiked with high (1.5 mg/dL) and low (0.15 mg/dL) levels of α-tocopherol. To estimate the effect on ion response at each concentration, average area of each set was used as follows:

The average suppression or enhancement should not exceed ± 25% and the coefficient of variation (% CV) for each concentration should be ≤15%. The stability of α-tocopherol and internal standard stock and working solutions was performed for three months at 4°C and six months at -20°C. Stability study of low (0.05 mg/dL) and high (2.00 mg/dL) concentrations of α-tocopherol in serum matrix was carried out after storing at room temperature (25°C for 24 hour), refrigeration temperature (4°C for 48 hours), and freezing temperature (-20°C for a month). After three cycles of freezing (-20°C) and thawing (25°C), freeze-thaw stability was assessed too. Stability of reconstituted extract was evaluated at 4°C for two days and at room temperature for half day. The effectiveness of the proposed method was checked by analysing eighty serum samples of patients submitted to the author’s laboratory. Samples were categorised into three groups based on age and each group is further sub-categorised into two sub-groups based on gender as presented in Table III to interpret the study findings. To the best of the author’s knowledge, the study to determine serum vitamin E levels in Pakistan has not yet been published.
RESULTS
The results of analytical method validation study are presented in Table II.
The linear range of 0.05–2.0 mg/dL for α-tocopherol in serum was obtained with ≥0.985 coefficient of linearity (r2). Established LLOQ and LOD were 0.05 mg/dL and 0.025 mg/dL, respectively. The accuracy at LLOQ was 100.1% with the precision ranged from 4.6-5.5%. The typical LC-MS/MS chromatogram of α-tocopherol and its calibration curve in serum matrix are presented in Figures 1 and 2, respectively.
After introducing analog interference, the recovery of α-tocopherol determined a range within 80–120% that reflected efficient selectivity of the author’s method. Furthermore, carry over was not observed even after analysing the highest concentration (2 mg/dL) during the entire study. The accuracy was determined 96.5 – 99.8% for the low, medium and high levels. The intra-day precision of low, medium, and high levels was 4.2-4.9% whereas, the inter-day precision determined was 5.0-5.9%. These results showed excellent reliability and reproducibility of this method in serum samples. At low (0.15 mg/dL) and high concentration (1.5 mg/dL), % ion suppression was -15.9% and -15.9% for α-tocopherol. The results obtained were within the acceptable range. α-tocopherol and internal standard stock, and working solutions can be stored well at 4°C for three months -20°C for six months, without any significant changes in their concentration if protected from light. The α-tocopherol stability in serum sample is less than that for neat standard solution and can be stable at 25°C for a day, 4°C for two days and, -20°C for a month. Little degradation had been observed following three consecutive freeze-thaw cycles. Additionally, after extraction and reconstitution, the extract showed good stability at 4°C for two days and ambient temperature in the auto-sampler for half day. The percentage observed changes in concentration were determined during the stability study are presented in Table II.
A validated method was applied to the serum samples of eighty patients in fasting state. Results range obtained for each age group and gender-based subgroup are presented in Table III.
DISCUSSION
Analysis of vitamin E (α-tocopherol) in biological fluids like serum is quite difficult due to the low-concentration in biological fluids, presence of proteins, lipids, and inorganic salts at high concentration in the matrix. An efficient extraction method is essential to eliminate the matrix interferences for reliable analysis. Various HPLC methods exist in literature,11,12,16-19 which are subjected to laborious sample preparation steps like saponification or expensive solid-phase extraction techniques to reduce the matrix interferences in vitamin E analysis. Most of the published LC-MS/MS methods has reported the determination of fat-soluble vitamins simultaneously, which is not cost-effective for clinical laboratories which receive test requests for vitamin E analysis only. Andreoli and colleagues did not use isotopic internal standards to compensate matrix interference in their study.20 Capote et al. simultaneously determined fat soluble vitamins with LC-MS/MS using a greater sample volume (1000 μL) following a solid phase extraction technique, which was unsuitable for routine tests in a clinical set up.21 Midttun and colleagues although, used an appropriate sample volume with faster analysis but they used chloroform for extraction which is carcinogenic.22 Similarly, Albaharani used a reduced sample volume with fast analysis time, but it required longer sample preparation process as compared to this study.13 In contrast, Ertugrul demonstrated analysis of fat-soluble vitamins simultaneously, using low sample volume but major limitations include longer analysis time and reduced sensitivity, probably due to matrix interferences as they used only protein precipitation technique for sample preparation.23
Table I: LC-MS/MS operating parameters for the estimation of vitamin E in serum.|
UHPLC parameters |
Value |
||||||
|
Injection volume (µL) |
5 |
||||||
|
Needle wash time (sec) |
20 |
||||||
|
Mobile phase A |
0.1% Formic acid in water |
||||||
|
Mobile phase B |
100%Methanol |
||||||
|
Gradient Elution Mode |
0.00 min |
75% B |
|||||
|
0.10 min |
100% B |
||||||
|
0.50 min |
100% B |
||||||
|
3.20 min |
100% B |
||||||
|
5.00 min |
75% B |
||||||
|
Stop time (min) |
5 |
||||||
|
Post time (min) |
2 |
||||||
|
MS/MS parameters |
Value (+) |
||||||
|
Ionization |
Air Jet stream (AJS) electron spray Ionization (ESI) |
||||||
|
Polarity |
Positive |
||||||
|
Gas temperature (oC) |
300 |
||||||
|
Sheath gas temperature (oC) |
325 |
||||||
|
Nitrogen gas flow (L/min) |
10 |
||||||
|
Nebulizer pressure (psi) |
40 |
||||||
|
Capillary voltage (V) |
4000 |
||||||
|
Nozzle voltage/ charging (V) |
1500 |
||||||
|
MRM transitions used |
|||||||
|
Compound |
Precursor ion |
Product ion |
Dwell time (ms) |
Fragmentor voltage (V) |
Collision Energy (V) |
||
|
Alpha-Tocopherol |
431.2 |
165.1 |
50 |
50 |
15 |
||
|
Alpha-Tocopherol |
431.2 |
83 |
50 |
50 |
25 |
||
|
Deuterated Alpha-Tocopherol (d6) IS |
437.5 |
171.1 |
50 |
50 |
5 |
||
Table II: Validation Summary for α-tocopherol determination in serum by LCMSMS.
|
Parameters |
Observed value |
|||||||||||||||
|
Limit of detection (LOD)* |
LOD conc. (mg/dL) |
Intra-day precision (% CV) |
Inter-day precision (% CV) |
|||||||||||||
|
0.025 |
5.3 |
5.2 |
||||||||||||||
|
Lower Limit of Quantitation (LLOQ)* |
LLOQ conc. (mg/dL) |
Intra-day precision (% CV) |
Inter-day Precision (% CV) |
Accuracy |
||||||||||||
|
0.05 |
5.5 |
4.6 |
100.1 |
|||||||||||||
|
Linearity and calibration curve** |
Linear range (mg/dL) |
Correlation coefficient (r2) |
Linear equation |
|||||||||||||
|
0.05-2.00 |
0.9908-0.9997 |
y = 109.673598 * x + 2.957161 |
||||||||||||||
|
Precision (% CV)* |
Intra-day Precision |
Inter-day Precision |
||||||||||||||
|
Low conc. (0.15 mg/dL) |
4.6 |
5.9 |
||||||||||||||
|
Mid conc. (0.75 mg/dL) |
4.9 |
5.4 |
||||||||||||||
|
High conc. (1.50 mg/dL) |
4.2 |
5.0 |
||||||||||||||
|
Accuracy (% Recovery)* |
Low conc. (0.15mg/dL) |
Medium conc. (0.75mg/dL) |
High conc. (1.50mg/dL) |
|||||||||||||
|
96.5 |
99.3 |
99.8 |
||||||||||||||
|
Ionization suppression |
Low conc. (0.15 mg/dL) |
High conc. (1.50 mg/dL) |
||||||||||||||
|
Relative standard deviation of analyte (%) |
5.0 |
2.0 |
||||||||||||||
|
Relative standard deviation of IS (%) |
2.0 |
1.0 |
||||||||||||||
|
Ionization suppression of analyte (%) |
-15.9 |
-15.2 |
||||||||||||||
|
Ionization suppression of IS (%) |
-14.1 |
-12.4 |
||||||||||||||
|
Stability study % Observed change in concentration |
||||||||||||||||
|
Stability of standard solutions |
α-tocopherol |
Internal Standard |
||||||||||||||
|
Stock solution at 4oC for 3 months |
-2.79 |
-1.66 |
||||||||||||||
|
Stock solution at -20oC for 6 months |
-1.70 |
-1.12 |
||||||||||||||
|
Working solution at 4oC for 3 months |
-3.19 |
-2.34 |
||||||||||||||
|
Working solution at -20oC for 6 months |
-2.30 |
-2.01 |
||||||||||||||
|
Three Freeze-Thaw Cycle |
At low conc. (0.05 mg/dL) |
At high conc. (1.50 mg/dL) |
||||||||||||||
|
-6.85 |
-4.44 |
|||||||||||||||
|
Stability in biological matrix |
25oC (24hr) |
4 oC (48hr) |
-20 oC (30 days) |
|||||||||||||
|
At low conc. (0.05mg/dL) |
-4.79 |
-3.46 |
-7.19 |
|||||||||||||
|
At high conc. (1.50 mg/dL) |
-6.58 |
-4.17 |
-5.10 |
|||||||||||||
|
Stability of reconstituted extract |
25oC (12hr) |
4 oC (48hr) |
||||||||||||||
|
At low conc. (0.05mg/dL) |
-3.88 |
-2.73 |
||||||||||||||
|
At high conc. (1.50 mg/dL) |
-4.01 |
-3.97 |
||||||||||||||
|
*Reported LOD/LLOQ, precision, and recovery values are based on fifteen determinations at each concentration. **Linear equation based on three determinations for each concentration of five concentrations per day for five days. |
||||||||||||||||
In this study, serum sample was used to extract vitamin E to minimise potential matrix interferences as compared to blood. During the method optimisation, the authors used high purity organic solvents to ensure compatibility with mass spectrometric analysis. Protein precipitation with organic solvent alone was not sufficient to reduce matrix interferences and resulted in reduced sensitivity; therefore, protein precipitation followed by liquid-liquid extraction technique was opted for. The reported sample preparation procedure was based on trying several options for the selection of appropriate buffer solution, precipitating solvent, extraction solvent, and then optimisation of volume of serum, and each solvent to ensure better recovery of vitamin E.
Table III: Application of validated method for determination of serum vitamin E by LCMSMS.|
Age (years) |
Gender (M=Male, F= Female) |
Sample size (n) |
Observed Serum Vitamin E level range (mg/dL) |
Optimal reference Range25 (mg/dL) |
Cut off value of vitamin E for deficiency (mg/dL) |
Interpretation based on findings |
|
≥1 to ˂12 |
M |
10 |
0.17-0.35 |
0.3-0.9 |
˂0.3 |
4 out of 10 males were vitamin E deficient |
|
F |
10 |
0.10-0.42 |
3 out of 10 females were vitamin E deficient |
|||
|
≥12 to ˂20 |
M |
10 |
0.38-0.73 |
0.6-1.0 |
˂0.6 |
7 out of 10 males were vitamin E deficient |
|
F |
10 |
0.18-0.63 |
5 out of 10 females were vitamin E deficient |
|||
|
≥20 to ˂85 |
M |
20 |
0.05-0.69 |
0.5-1.8 |
˂0.5 |
16 out of 20 males were vitamin E deficient |
|
F |
20 |
0.07-0.59 |
12 out of 20 females were vitamin E deficient |
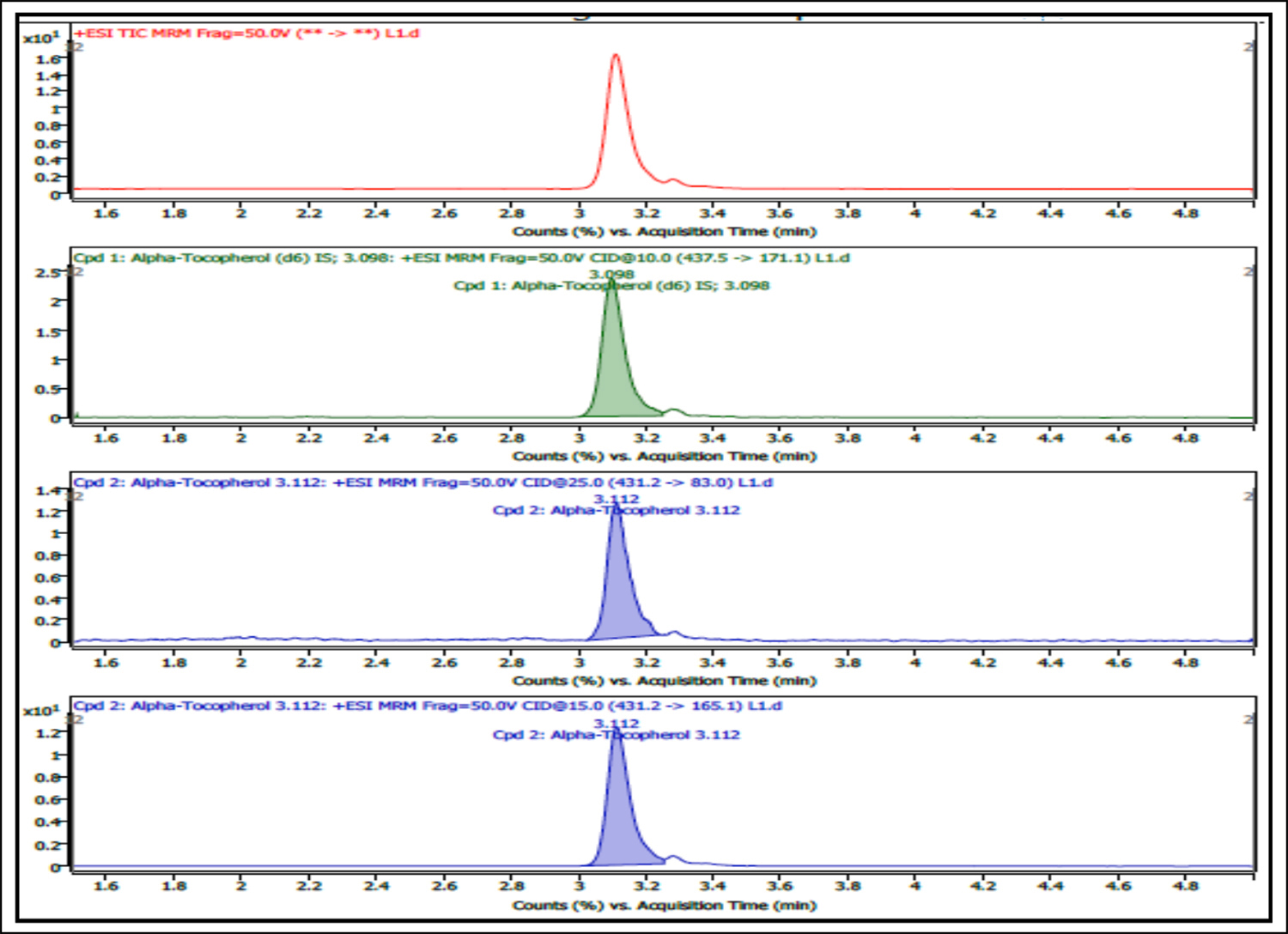 Figure 1: Typical LC-MS/MS total ion chromatogram of alpha-tocopherol in serum (0.05mg/dL).
Figure 1: Typical LC-MS/MS total ion chromatogram of alpha-tocopherol in serum (0.05mg/dL).
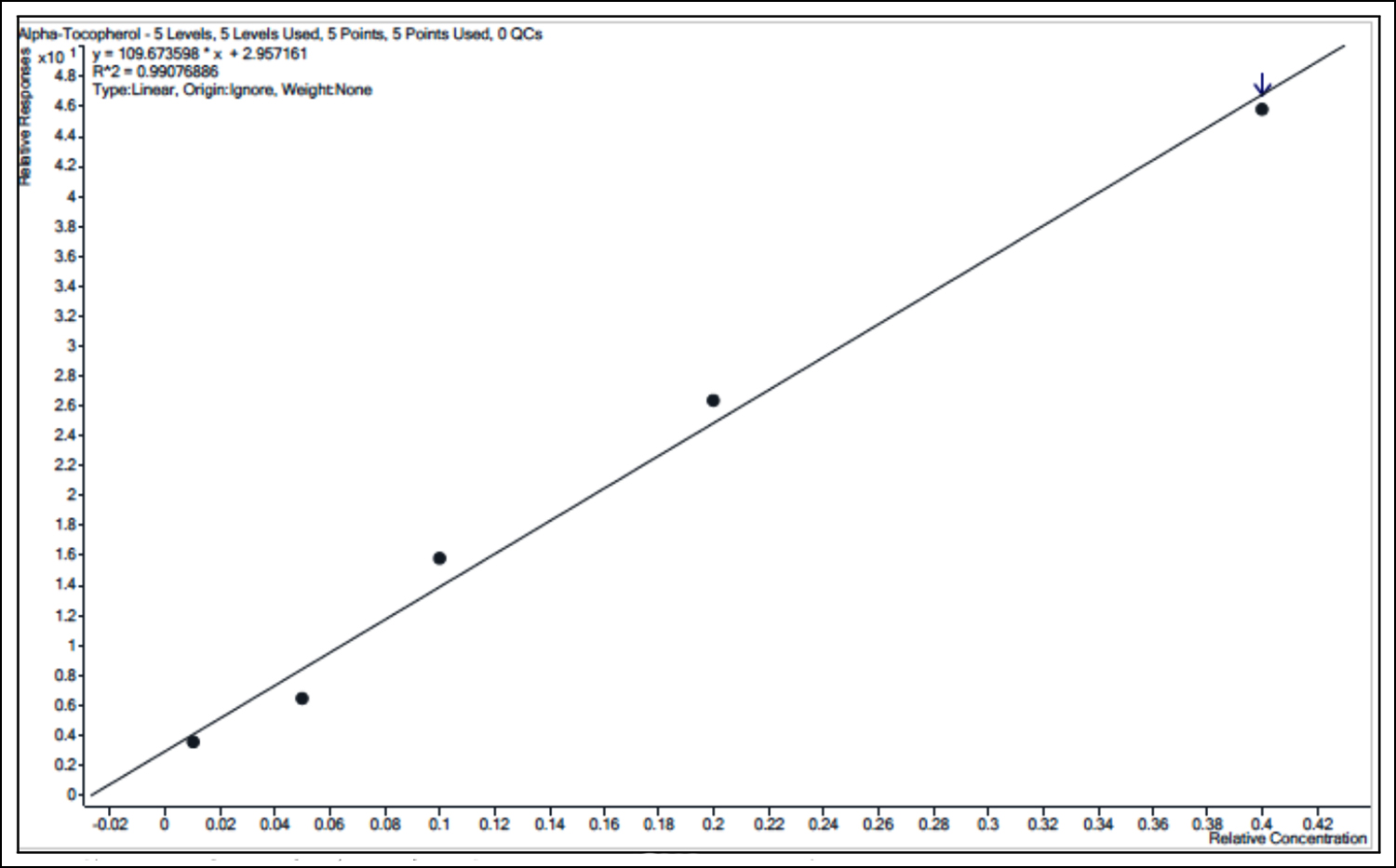 Figure 2: Linearity curve for alpha-tocopherol in serum (0.05 - 2.0mg/dL).
Figure 2: Linearity curve for alpha-tocopherol in serum (0.05 - 2.0mg/dL).
Methanol was selected to precipitate protein because it was used to prepare in-house calibrators/controls (including isotopic internal standard), compatible with hexane and LC-MS/MS analysis. Hexane is widely used as extraction solvent in literature for vitamin analysis11,24 as it is immiscible with water and less dense than water and methanol. Due to these physical properties and better recovery of vitamin E as compared to other extraction solvents, hexane became the potential extraction solvent in this study. The finalised extraction method showed good agreement with recommendations for vitamin E analysis based on literature evidences.12 The most crucial step in method development is to prepare matrix matched in-house calibrators and controls to ensure compatibility. In vitamin E analysis, the actual issue was faced by analysts is to find suitable matrix which is free of endogenous vitamin E. The authors of this study used SeraCon-DL for this purpose, although it contained endogenous 25-OHD3 but it showed no interference with vitamin E analysis.
Reversed-phase chromatography was used in this study. To ensure better chromatographic separation, many columns were tried but best separation was achieved on the Agilent Infinity-Lab Poroshell 120EC-C18; 2.1mm x 50mm x 2.7 µm chromatographic column. Similarly, to eliminate co-elution of phospholipids and better resolution, various mobile phase gradient elution profile, flow rate, injection volume, and column temperature were investigated during this study. MS/MS parameters were optimised using vitamin E solution in methanol and serum samples. The most commonly used ionisation mode in literature for vitamin testing is based on atmospheric pressure chemical ionisation but the authors of the current study demonstrated better results using electron spray ionization (ESI) operated in positive-ion mode and multiple-reaction monitoring (MRM). Single-ion transition for isotopic internal standard and two ions (quantifier and qualifier) transitions for α-tocopherol were utilised.
The authors of this study have reported a sensitive as well as selective tandem mass spectrometric method for α-tocopherol (vitamin E) analysis using low sample volume (100 μL) was developed. The studied method demonstrated excellent performance pertaining to the precision, accuracy, and reportable range for vitamin E. The authors tried to fill the analytical and knowledge gaps in existing studies. The proposed technique was successfully applied to quantitate vitamin E in serum specimens of eighty patients. 67.5% males and 50% females were found vitamin E deficient, but these values need to be further verified by increasing sample size and elaborating criteria for sample selection for future studies. Extended study to determine the status of serum vitamin E in native population was beyond the scope of the author’s current study and will be carried out specifically in the future. This method has been extensively validated and is reproducible in clinical or diagnostic laboratories for patient sample testing.
CONCLUSION
The studied method is an efficient, reliable, and precise method for vitamin E analysis in serum samples. Low sample volume utilisation is an ideal condition for clinical laboratories especially, when multiple tests have to be performed and submitted sample size is limited. Use of six sites deuterated internal standard, simple extraction steps, better extraction recovery and extensive gradient elution to further minimise interferences have made this method a suitable candidate for utilisation as reference method.
ETHICAL APPROVAL
An ethical approval was taken from the institutional review board of Chughtai Institute of Pathology, Lahore.
PATIENTS CONSENT
Informed consent was taken from all the study participants.
COMPETING INTEREST:
The authors declared no competing interest.
AUTHORS’ CONTRIBUTION:
HSM: Substantial contribution to the conception or design of the work, or the acquisition, analysis or interpretation of data for the work.
MDK: Revising the work critically for important intellectual content.
ORC: Final approval of the version to be published.
ZB, AA, HB: Acquisition and analysis.
All the authors have approved the final version of the manuscript to be published.
REFERENCES
- Galli F, Bonomini M, Bartolini D, Zatini L, Reboldi G, Marcantonini G, et al. Vitamin E (Alpha-Tocopherol) Metabolism and Nutrition in Chronic Kidney Disease. Antioxidants 2022; 11(5):989. doi: 10.3390/antiox11050989.
- Regina Brigelius-Flohé. Vitamin E research: Past, now and future. Free Radical Biol Med 2021; 177:381-90. doi: 10.1016/j.freeradbiomed.2021.10.029.
- Panel on dietary antioxidants and related compounds, subcommittees on upper reference levels of nutrients and interpretation and uses of DRIs, standing committee on the scientific evaluation of dietary reference intakes, food and nutrition board, institute of medicine, dietary reference intakes for vitamin C, vitamin E, selenium, and carotenoids. National Academy Press; Washington DC: 2000.
- Hardman JG, Limbird LE, Gilman AG. Goodman & Gilman's The Pharmacological Basis of Therapeutics. Ed.10th, New York: McGraw-Hill; 2001.
- Ford ES, Ajani UA, Mokdad AH. Brief communication: The prevalence of high intake of vitamin E from the use of supplements among U.S. adults. Ann Intern Med 2005; 143(2):116-20. doi: 10.7326/0003-4819-143-2-2005071 90-00010.
- Miller ER, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ and Guallar E. Meta-analysis: High-dosage vitamin E supplementation may increase allcause mortality. Ann Intern Med 2005; 142(1): 37-46. doi: 10.7326/0003-4819- 142-1-200501040-00110.
- Albahrani AA, Rotarou V, Roche PJ and Greaves RF. Analyte stability during the total testing process: Studies of vitamins A, D and E by LC-MS/MS. Clin Chem Lab Med 2016; 54[10]: 1609-18.
- Danilo G, Roberta G, Carolina B, Pierangelo T, Desirée B, Simone M, et al. LC-MS/MS assay for the simultaneous determination of tocopherols, polyunsaturated fatty acids and their metabolites in human plasma and serum. Free Radar Bio Med 2019; 144:134-43. doi: 10.1016/j.freeradbiomed. 2019.04.017.
- Drammeh BS, Schleicher RL, Pfeiffer CM, Jain RB, Zhang M, Nguyen PH. Effects of delayed sample processing and freezing on serum concentrations of selected nutritional indicators. Clin Chem 2008; 54(11):1883-91. doi: 10.1373/clinchem.2008.108761.
- Clark S, Youngman LD, Chukwurah B, Palmer A, Parish S, Peto R, et al. Effect of temperature and light on the stability of fat-soluble vitamins in whole blood over several days: implications for epidemiological studies. Int J Epidemiol 2004; 33(3):518-25. doi: 10.1093/ije/dyh064.
- Greaves R, Jolly L, Woollard G, Hoad K. Serum vitamin A and E analysis: Comparison of methods between laboratories enrolled in an external quality assurance programme. Ann Clin Biochem 2010; 47 (Pt 1):78-80. doi: 10.1258/acb. 2009.009116.
- Greaves RF, Woollard GA, Hoad KE, Walmsley TA, Johnson LA, Briscoe S, et al. Laboratory medicine best practice guideline: vitamins A, E and the carotenoids in blood. Clin Biochem Rev 2014; 35(2):81-113.
- Albahrani AA, Rotarou V, Roche PJ, Greaves RF. A simultaneous quantitative method for vitamins A, D and E in human serum using liquid chromatography-tandem mass spectrometry. Steroid Biochem Mol Biol 2016; 159:41-53. 107. doi: 10.1016/j.jsbmb.2016.02.019.
- Pitt JJ. Principles and applications of liquid chromatography-mass spectrometry in clinical biochemistry. Clin Biochem Rev 2009; 30(1):19-34.
- Vogeser M, Seger C. Pitfalls associated with the use of liquid chromatography-tandem mass spectrometry in the clinical laboratory. Clin Chem 2010; 56(8):1234-44. doi: 10.1373/clinchem.2009.138602.
- Alvarez JC, Demazancourt P. Rapid and sensitive high-performance liquid chromatographicmethod for simultaneous determination of retinol, alpha-tocopherol, 25-hydroxyvitamin D3 and25-hydroxyvitamin D2 in human plasma with photodiode-array ultraviolet detection. J Chromatogr B Biomed Sci Appl 2001; 755(1-2):129-35.
- Mata-Granados JM, Luque De Castro MD, Quesada JM. Fully automated method for the determination of 24, 25(OH)D2 and 25(OH) D3 hydroxyvitamins, and vitamins A and E in human serum by HPLC. J Pharm Biomed Anal 2004; 35(3):575-82. doi: 10.1016/j.jpba.2004.01.027.
- Quesada JM, Mata-Granados JM, Luque De Castro MD. Automated method for the determination of fat-soluble vitamins in serum. J Steroid Biochem Mol Biol 2004; 89-90(1-5): 473-7. doi: 10.1016/j.jsbmb.2004.03.056.
- Zhang H, Quan L, Pei P, Lin Y, Feng C, Guan H, et al. Simultaneous determination of Vitamin A, 25-hydroxyl vitamin D3 α-tocopherol in small biological fluids by liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2018; 1079:1-8. doi: 10.1016/ j.jchromb.2017.12.017.
- Andreoli R, Manini P, Poli D, Bergamaschi E, Mutti A, Niessen WM. Development of a simplified method for the simultaneous determination of retinol, alpha-tocopherol, and beta-carotene in serum by liquid chromatography-tandem mass spectrometry with atmospheric pressure chemicalionization. Anal Bioanal Chem 2004; 378(4):987-94. doi: 10.1007/ s00216-003-2288-0.
- Priego Capote F, Jiménez JR, Granados JM, de Castro MD. Identification and determination of fat-soluble vitamins and metabolites in human serum by liquid chromatography/triple quadrupole mass spectrometry with multiple reaction monitoring. Rapid Commun Mass Spectrom 2007; 21(11):1745-54. doi: 10.1002/rcm.3014.
- Midttun O, Ueland PM. Determination of vitamins A, D and E in a small volume of human plasma by a high‐throughput method based on liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom 2011; 25(14): 1942-8. doi: 10.1002/rcm.5073.
- Ertugrul S, Yucel C, Sertoglu E, Ozkan Y, Ozgurtas T, Development and optimization of simultaneous determinatıon of fat soluble vitamins by liquid chromatography tandem mass spectrometry. Chemistry Physics Lipids 2020; 230:104932. doi.org/10.1016/j.chemphyslip.2020.104932.
- Luque-Garcia JL and Luque de Castro MD. Extraction of fat-soluble vitamins. J Chromatogr A 2001; 935(1-2):3-11. doi: 10.1016/s0021-9673(01)01118-9.
- Roberts NB. Taylor A. Sodi R: Chapter 37 Vitamins and Trace Elements. In Tietz Textbookof Clinical Chemistry and Molecular Diagnostics. Edited by N Rifai, AR Horvath, CT Wittwer. ed 6th. St. Louis, MO. Elsevier, 2018. pp 639-718.