Haematopoietic Stem Cell Transplantation (HSCT) for Primary Immune System Disorders in Children: A Single Centre Experience
By Asghar Ali Kerio, Tariq Azam Khattak, Tariq Ghafoor, Muhammad Yousaf, Nighat Shahbaz, Qamar Un Nisa Chaudary
Affiliations
doi: 10.29271/jcpsp.2023.03.341ABSTRACT
Objective: To determine the outcomes of allogeneic HSCT in children with primary immune system disorders (PID).
Study Design: Descriptive Cross-sectional study.
Place and Duration of the Study: Armed Forces bone marrow transplant centre / National Institute of Bone Marrow Transplant (AFBMTC / NIBMT), Rawalpindi, Pakistan, from October 2012 to December 2021.
Methodology: Data of all cases undergoing HSCT for immune system disorders were analysed for variables affecting outcome and overall survival in the first 180 days after allogeneic HSCT. All patients presenting to AFBMTC / NIBMT with PID, age <12 years. Patients with organ dysfunction secondary to repeated infections were excluded from the study. Data of all patients and their donors undergoing HSCT for immune system disorders were analysed for variables affecting outcome and overall survival in the first 180 days after allogeneic bone marrow transplant. Neutrophil engraftment was defined as absolute neutrophil count ≥0.5 × 109/L for 3 consecutive days, while platelet engraftment as platelet count ≥20 × 109/L without platelet transfusion for one week. Overall survival (OS) was taken as time from the date of HSCT till day + 180 post-transplant.
Results: A total of 42children including 29 boys and 13 girls underwent HSCT for PID. The mean age was 2.1±2.8 years. Underlying diagnosis was haemophagocytic lymphohistiocytosis (HLH), severe immune deficiency (SCID), leukocyte adhesion defect (LAD), X-linked agammaglobulinemia, chronic granulomatous disease (CGD) and Job’s syndrome in 18 (42.9%), 16 (38.1%), 3(7.1%), 2 (4.8%), 2 (4.8%) and 1 (2.4%) patients respectively. Thirty-one (73.8%) children had fully HLA-matched donors while 11 (26.2%) had haplo-matched donors. Major immediate post-transplant complications were febrile neutropenia, mucositis and SOS/VOD in 31 (73.8%), 9 (21.4%) and 4 (10.0%) cases, respectively. Eight (19.0%) had CMV reactivation, acute GVHD was seen in 17 (40.4%) cases, while 1 (2.3%) case had chronic GVHD. Twelve (28.6%) patients died, out of which 5 had graft failure, 3 had VOD, 2 had pneumonia, 1 had severe GVHD, and 1 died due to seizures. Overall survival (OS) in this study was 71.4% with survival reaching up to 80.6% in fully matched HSCT.
Conclusion: HLH and SCID were the commonest immune disorders requiring HSCT. Graft failure leading to neutropenic sepsis was the commonest cause of mortality. OS was better in fully matched HSCT as compared to haplo-identical HSCT.
Key Words: Immune deficiency, Severe combined immunodeficiency, Haematopoietic stem cell transplantation.
INTRODUCTION
Primary immune deficiency (PID) refers to a heterogeneous group of disorders characterised by poor or absent function in one or more components of immune system which predispose affected individuals to increase frequency and severity of infection.1
Genetically inherited disorders of the immune system expose the affected children to the higher risk of infection, inflammation, and autoimmunity.2 Replacement of the defective recipient immune system with the functional system from a healthy donor, by haematopoietic stem cell transplantation (HSCT), results in a permanent cure for some of these disorders.3 PID has seen a lot of revolutionary breakthroughs in the last 20 years including advancement in HLA-tissue typing methods, adaptation of conditioning regimen from amyeloablative regimen to reduced intensity conditioning regimen (RIC) as myeloablative regimen is associated with high transplant-related mortality (TRM), accessibility of alternative stem cell sources, improved GVHD prophylaxis, and better infection control strategies.4
Allogeneic HSCT in Pakistan has been conducted over about three decades now, at various centres.5 There is no formal data available regarding the outcome of HSCT in PID. This study was conducted to determine the outcome of allogenic HSCT in children suffering from primary immune system disorders at our centre.
METHODOLOGY
After the approval from the institutional review board, informed/written consents were sought from parents/guardians of all study participants for the confidentiality of the data for this study. This descriptive study was conducted at The Armed Forces Bone Marrow Transplant Centre/ National Institute of Bone Marrow Transplant (AFMBTC /NIBMT) Rawalpindi, Pakistan from October 2012 to December 2021.
All patients (both genders, age: <12 years) presenting to AFBMTC/NIBMT with PID were included in the study. While patients with organ dysfunction secondary to repeated infections were excluded from the study.
Neutrophil engraftment was defined as absolute neutrophil count ≥0.5 × 109/L for 3 consecutive days, while platelet engraftment as platelet count ≥20 × 109/L without platelet transfusion for one week. Primary graft failure was defined as failure to achieve an ANC of 0.5 × 109/L by day 28 and the secondary graft failure as ANC <0.5 × 109/L or donor chimerism less than 10% by molecular analysis in patients who previously achieved an ANC of 0.5 x 109/L. Overall survival (OS) was taken as time from the date of HSCT till the day + 180 post-transplant.
Data of all patients and their donors undergoing HSCT for the immune system disorders were analysed for variables affecting outcome and overall survival in the first 180 days, after allogeneic bone marrow transplant. Variables were age, gender, diagnosis of underlying immune disorder, donor characteristics, conditioning protocol, source and dose of stem cell, days of neutrophil, and platelets engraftment and post-transplant complications. Acute Grafte vs Host Disease (GVHD) was classified according to modified Glucksberg staging,6 as shown in Table I.
Data was entered in pre-designed proforma and was analysed using SPSS version 26.0. Mean and standard deviation were calculated for the quantitative variables like age of the patients and donors, stem cell dose, neutrophil and platelets engraftment days. Frequency and percentages were calculated for qualitative variables like the gender of patient and donor, disease diagnosis, HLA typing (full matched /Haplo-matched), relation of the donor with patient and post-transplant complications. Kaplan-Meier survival analysis was used. Statistical significance (Log-Rank) was defined as a p-value <0.05.
RESULTS
During this study period, a total of 42 children underwent HSCT for PID. Patients characteristics are shown in Table I.
Table II is showing the characteristics of donors (gender, age, relation with patient and ABO grouping). The mean age of the donors was 17.9 ±13.2 years while 25 (25.9%) donors were females. The most common donor relation was sister 16 (38.1%) and father 9 (21.4%).
All patients received RIC protocol consisting of Fludarabine, Busulfan and Thymoglobulin. Three patients with SCID had an initial stem cells infusionwithout conditioning followed by transplant with conditioning. All patients received Cyclosporine and Mycophenolate mofetil for GVHD prophylaxis.Bone marrow was the source of stem cells in all cases. Three cases also received peripheral stem cells (PBSC) in addition to BM to achieve adequate stem cell dose. Mean total nucleated cells (TNC) and CD 34 dose were 6.42±2.75x108/Kg and 9.3±7.0x106/Kg, respectively. The mean neutrophil and platelets engraftment days were 13.83±2.69 and 21.91±10, respectively.
Neutropenic fever was documented in 31 (73.8%) cases. Acute GVHD was seen in 17 (40.47%) patients. Fifteen (88.2%) patients had only skin GVHD (10 had stage I, 3 had stage II, and 2 had stage III skin GVHD). One patient had both gut and skin GVHD (grade II) while one patient who had haplo-BMT with father, had severe GVHD involving skin, gut, and liver (grade III) resulting in mortality. One (2.3%) patient had chronic skin GVHD. Gender disparity between patient and donor resulted in statistically significant high incidence of GVHD (p-value 0.01). Donor age >10 years has also statistically significant association with the high rate of GVHD (p-value 0.002). Nine (21.4%) cases developed oral mucositis. Two patients had grade II mucositis, while one patient had grade III mucositis. Seven (16.6%) cases including, 3 HLH and 2 each of Agammaglobulinemia and LAD had graft failure. CMV reactivation was documented in 8 (19.0%) patients. Sinusoidal obstructive syndrome (SOS/VOD) was diagnosed in 4 (10.0%) patients. The average time of onset of VOD was 10 days.
Twelve (28.6%) cases expired, out of which6 (50%) had Haplo-identical HSCT. The cause of mortality was graft failure followed by neutropenic sepsis in 5 (45.4%) cases. Three (27.7%) cases suffering from SCID who had Haplo-BMT died of VOD. Two patients died due to severe respiratory infection (fungal pneumonia), while 1 death was due to the grade III acute GVHD in Haplo-BMT. One case died of aspiration pneumonia secondary seizures.
Overall survival (OS) in this study at day 180 was 71.4%. Disease-wise OS is given in Figure 3. OS in fully matched versus haplo-matched HSCT was 80.6% and 45.5%, respectively (p-value of <0.002), as shown in Figure 2. Disease-free survival was 64.3% as shown in the Kaplan-Meier survival curve below.
DISCUSSION
Most of the immune disorders carry a high risk of morbidity and mortality. Children without definitive treatment usually die within the first year of life. Currently, HSCT is the only curative option in these disorders as gene therapy is still in the experimental phases.7 In this study, the authors are reporting the largest cohort of PID who had HSCT in Pakistan.
Table I: Glucksberg staging and grading.
|
Organ |
Stage |
Description |
|
Skin |
1 |
Maculopapular rash over <25% of body area |
|
2 |
Maculopapular rash over 25 to 50% of body area |
|
|
3 |
Generalised erythroderma |
|
|
4 |
Generalised erythroderma with bullous formation and often with desquamation |
|
|
Liver |
1 |
Bilirubin 2.0 to 3.0 mg/dL |
|
2 |
Bilirubin 3.1 to 6.0 mg/dL |
|
|
3 |
Bilirubin 6.1 to 15.0 mg/dL |
|
|
4 |
Bilirubin >15.0 mg/dL |
|
|
Gut |
1 |
Diarrhoea >30 mL/kg or >500 mL/day |
|
2 |
Diarrhoea >60 mL/kg or >1000 mL/day |
|
|
3 |
Diarrhoea >90 mL/kg or >1500 mL/day |
|
|
4 |
Diarrhoea >90 mL/kg or >2000 mL/day; or severe abdominal pain with or without ileus |
|
|
Glucksberg grading |
||
|
Grade |
Description |
|
|
I |
Stage 1 or 2 skin involvement; no liver or gut involvement; ECOG PS 0 |
|
|
II |
Stage 1 to 3 skin involvement; Grade 1 liver or gut involvement; ECOG PS 1 |
|
|
III |
Stage 2 or 3 skin, liver, or gut involvement; ECOG PS 2 |
|
Table II: Characteristics of patients (n=42).
|
Patients characteristics |
Number (%) |
|
|
Gender |
Male |
29 (69.0%) |
|
Female |
13 (31.0%) |
|
|
Age in years |
≤1 |
23 (54.9.0%) |
|
>1 |
19 (45.5%) |
|
|
Mean age |
2.1±2.8 years |
|
|
Range |
1-14 years |
|
|
Diagnosis |
Haemophagocytic Lymphohistiocytosis (HLH) |
18 (42.9%) |
|
Severe Immune Deficiency (SCID) |
16 (38.1%) |
|
|
Leukocyte Adhesion Defect (LAD) |
3 (7.1%) |
|
|
Agammaglobulinemia |
2 (4.8%) |
|
|
Chronic granulomatous Disease (CGD) |
2 (4.8%) |
|
|
Job’s Syndrome |
1 (2.4%) |
|
Table III: Characteristics of donors (n=42).
|
Donors characteristics |
Number (%) |
|
|
Gender |
Male |
17 (40.5%) |
|
Female |
25 (59.5%) |
|
|
Age in years Mean Range |
<10 |
18 (42.9%) |
|
10-20 |
6 (14.4%) |
|
|
20-30 |
7(16.8%) |
|
|
30-40 |
11 (26.4%) |
|
|
Relation with the patients |
Sister |
16 (38.1%) |
|
Brother |
8(19.0%) |
|
|
Father |
9 (21.4%) |
|
|
Mother |
8 (19.0%) |
|
|
Maternal Aunt |
1 (2.4%) |
|
|
ABO Mismatch |
No ABO Mismatch |
27(64.3%) |
|
Minor Mismatch |
08(19.0%) |
|
|
Major Mismatch |
07(16.7%) |
|
|
HLA matching |
Fully Matched |
31 (73.80%) |
|
Haplo Matched |
11 (26.2%) |
|
Mean age at the time of HSCT was 2.1±2.8 years in this study. Recent multicentre regional data from India also reported median age around 12 months, which are very similar to the findings of this study.8 The most common diagnosis among patients undergoing HSCT for immune disorders in this study was HLH followed by SCID. Raj et al. from India reported equal frequency (25%) of HLH and SCID among the children with PID.8
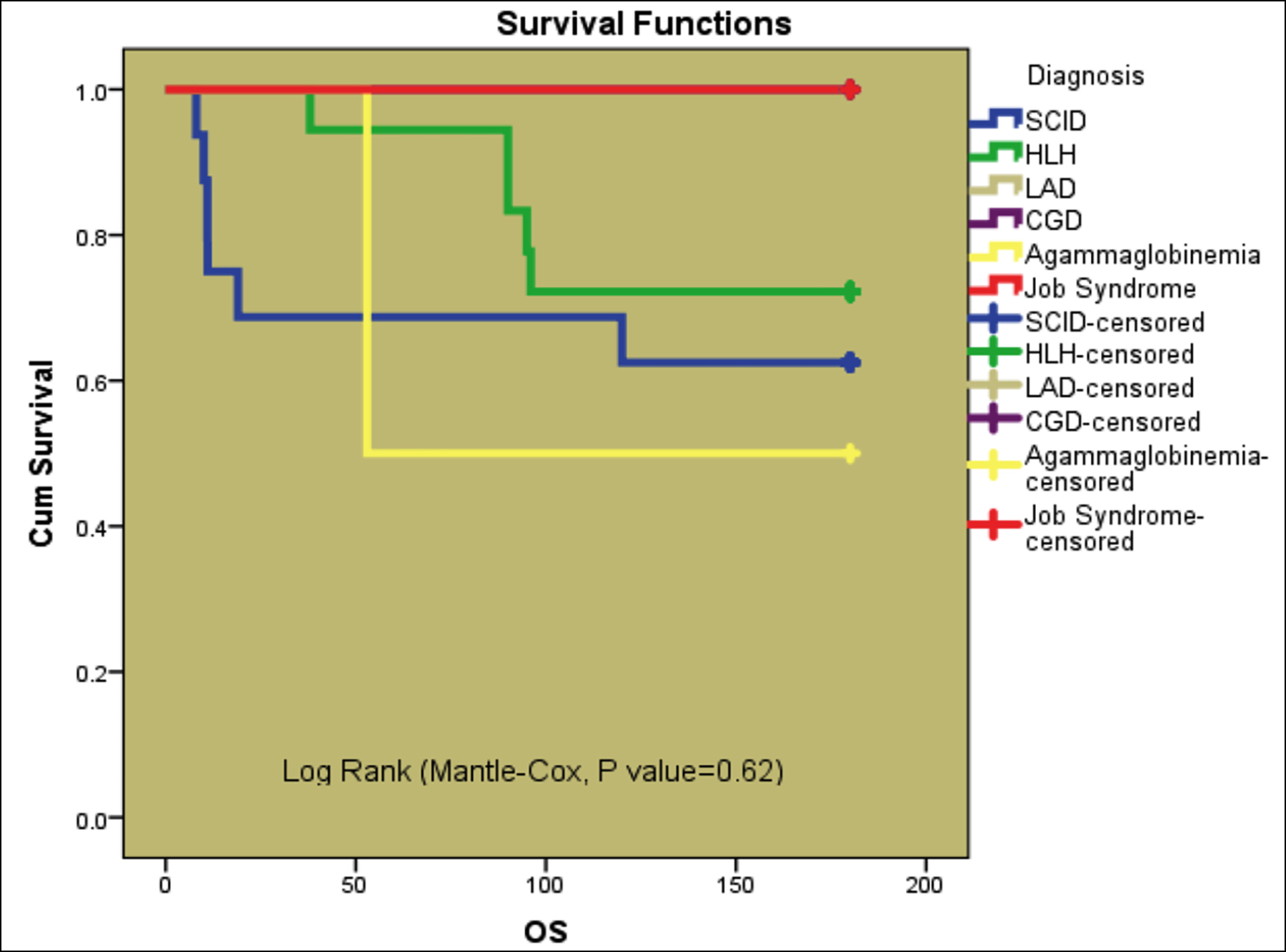 Figure 1: Overall survival: disease-wise.
Figure 1: Overall survival: disease-wise.
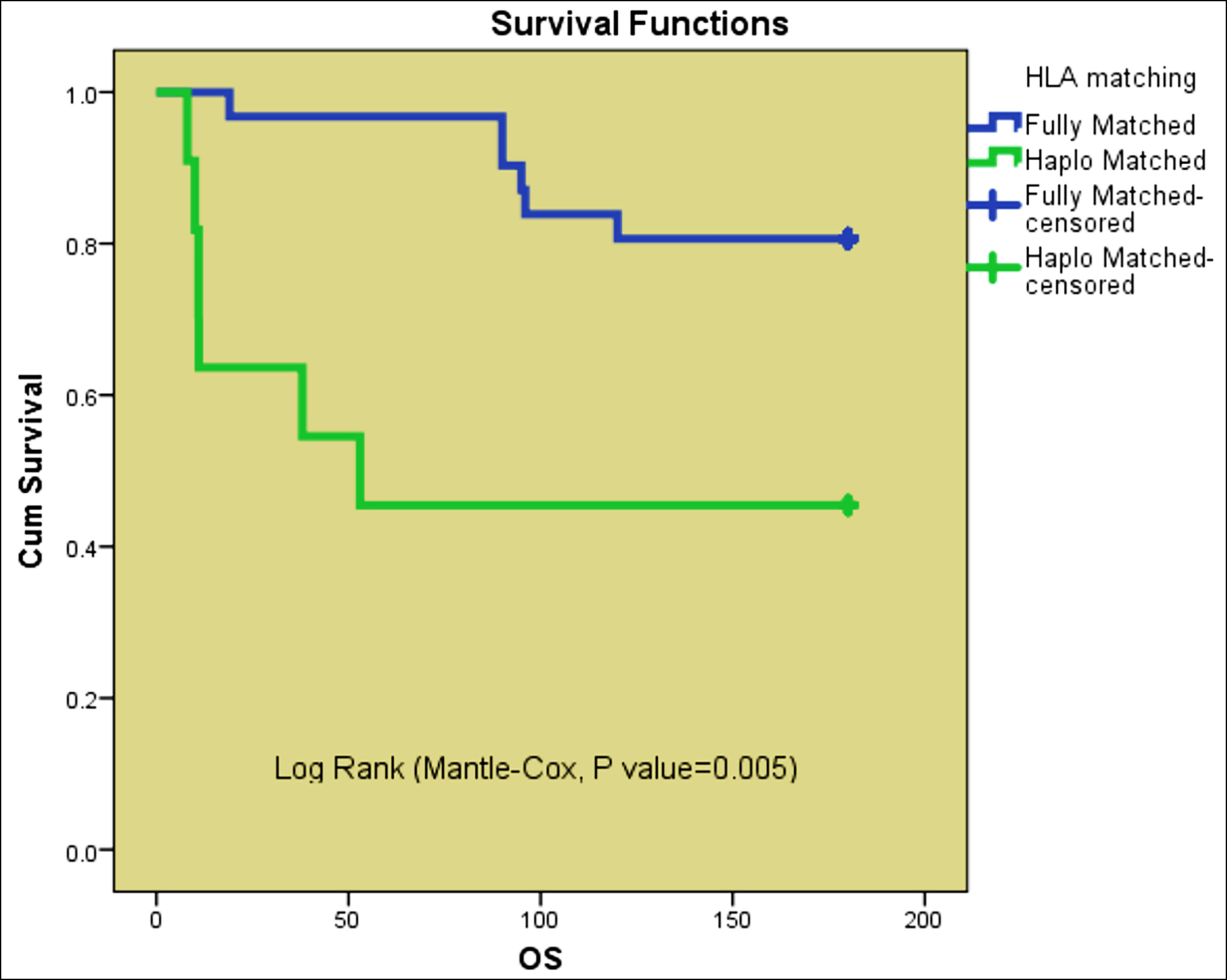 Figure 2: Overall survival: Fully matched vs. Haplo-Matched.
Figure 2: Overall survival: Fully matched vs. Haplo-Matched.
Overall, survival in the study was 71.4%.In the developed world, OS has improved from 65-70%9-11 to 85-90%12,13 and in recent years, a study conducted in Australia showed an OS 82-94%.14
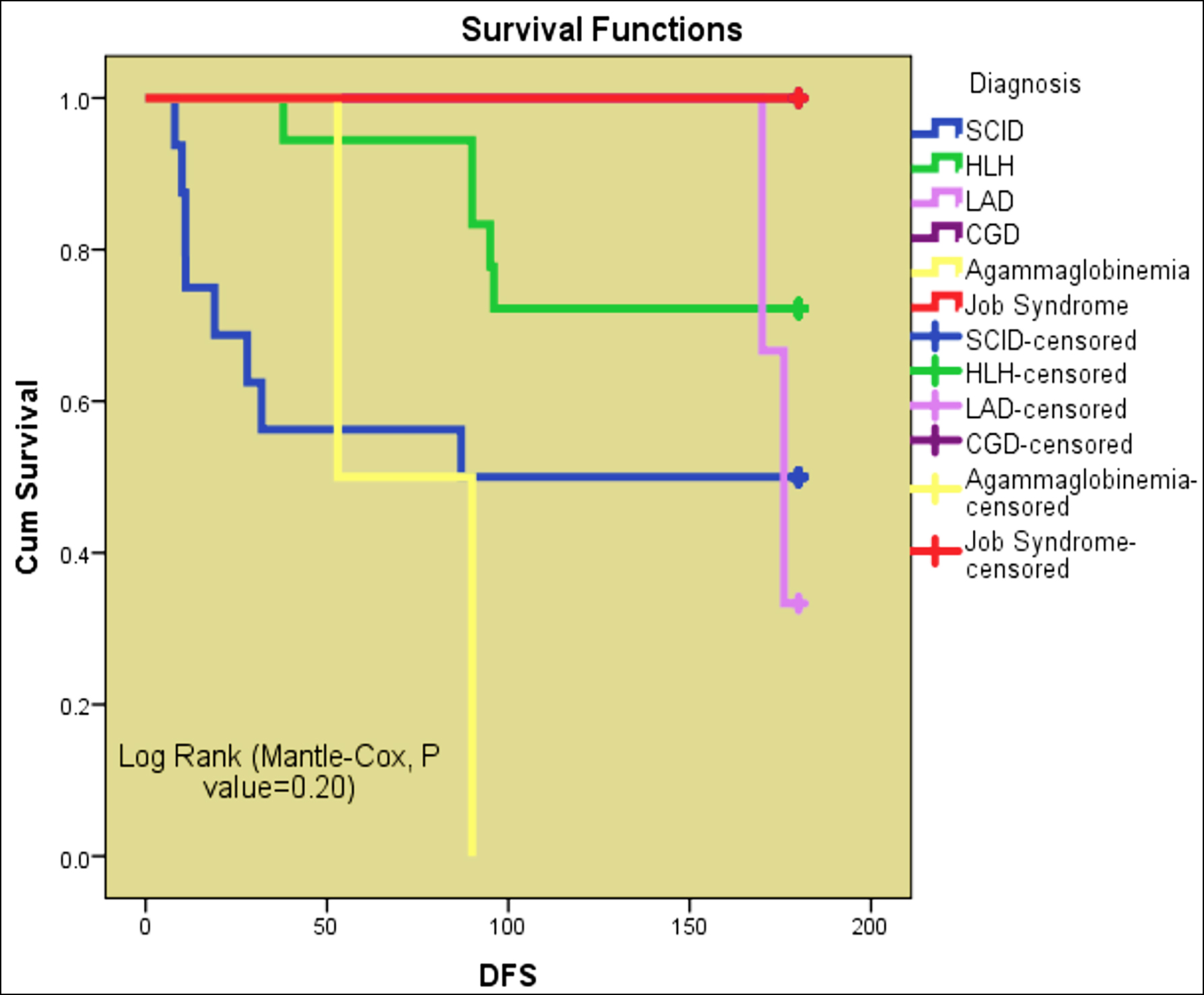 Figure 3: Disease-free survival.
Figure 3: Disease-free survival.
However, the data from developing countries reported survival rates between 65-70% which is quite similar to what the authors have documented.15,16 This difference in survival is due to the lack of supportive care, unavailability of screening programs for early diagnosis and poor management of infectious complications in these immune disorders. OS in MSD was almost double than in haplo-HSCT (80.6 vs 45.5%). An Indian study also reported better outcomes (OS 78% vs.61%) in fully matched vs haplo-HSCT.17In this study, 45.6% deaths were attributed to graft failure complicated by infections. Regional data from the India has shown that the infection was the commonest cause of death following HSCT for PID.17
This study had some limitations as well. As this was a single centre study with a relatively small sample size, the study’s findings cannot be generalised. Retrospective design of this study was another limitation. Being a rare group of disorders, a number of immunodeficiency patients undergoing HSCT at one centre will always remain low. In order to formulate a better HSCT strategy, it is suggested that all the HSCT centres in Pakistan analyse their data under the umbrella of recently established Pakistan Blood & Marrow Transplant (PBMT) group.
CONCLUSION
The authors concluded from this study that HLH and SCID were the commonest immune disorders requiring HSCT. Overall survival was superior in fully matched HSCT as compared with haplo-Identical HSCT. Graft failure followed by infections was the most common cause of mortality.
ETHICAL APPROVAL:
After the Approval from the institutional review board, informed/written consent were sought from parents/guardians of all study participants for the confidentiality of the data for this study (IRB/FCPS-025/AFBMTC/approval/2022).
PATIENTS’ CONSENT:
Informed written consent were taken from all patients.
COMPETING INTEREST:
This study has no competing interest to be declared by any author.
AUTHORS’ CONTRIBUTION:
AAK: The acquisition and drafting the work.
TKA: Conception and design of the work.
TG, NS: Revision of the work for important intellectual content.
MY: Analysis and interpretation of data for the work.
QNC: Final approval of the version to be published.
All the authors have approved the final version of the manuscript to be published.
REFERENCES
- Marshall JS, Warrington R, Watson W, Kim HL. An introduction to immunology and immunopathology. Allergy Asthma Clin Immunol 2018; 14(2):1-0. doi: 10.1186/s13223- 018-0278-1.
- Przepiorka D, Weisdorf D, Martin P, Klingemann HG, Beatty P, Hows J, et al. Consensus conference on acute GVHD grading. Bone Marrow Transplant 1995; 15(6): 825-8.
- Haddad E, Hoenig M. Hematopoietic Stem Cell Transplantation for Severe Combined Immunodeficiency (SCID). Front Pediatr 2019; 7:481.
- CastagnolI R, Delmonte OM, Calzoni E, Notarangelo LD. Hematopoietic stem cell transplantation in primary immunodeficiency disease: Current status and future perspectives. Front Pediatr 2019; 7:295. doi: 10. 3389/fped. 2019.00295.eCollection 2019.
- Shamsi TS, Hashmi K, Adil S, Ahmad P, Irfan M, Raza S, et al. The stem cell transplant program in Pakistan—the first decade. Bone Marrow Transplant 2008; 42(Suppl 1):S114-S117. doi: 10.1038/bmt.2008.137.
- Schoemans HM, Lee SJ, Ferrara JL, Wolff D, Levine JE, Schultz KR, et al. EBMT−NIH−CIBMTR Task Force position statement on standardized terminology & guidance for graft-versus-host disease assessment. Bone Marrow Transplant 2018; 53(11):1401-15. doi: 10.1038/s41409-018- 0204-7.
- Angural A, Spolia A, Mahajan A, Verma V, Sharma A, Kumar P, et al. Review: Understanding rare genetic diseases in Low Resource Regions like Jammu and Kashmir - India. Front Genet 2020; 11:415. doi:10.3389/fgene.2020.00415
- Raj R, Aboobacker FN, Yadav SP, Uppuluri R, Bhat S, Choudhry D, et al. Multicenter outcome of hematopoietic stem cell transplantation for Primary Immune Deficiency Disorders in India. Front Immunol 2021; 11:606930.
- Haddad E, Logan BR, Griffith LM, Buckley RH, Parrott RE, Prockop SE, et al. SCID genotype and 6-month posttransplant CD4 count predict survival and immune recovery. Blood 2018; 132:1737-49
- Pai SY, Logan BR, Griffith LM, Buckley RH, Parrott RE, Dvorak CC, et al. Transplantation outcomes for severe combined immunodeficiency, 2000-2009. N Engl J Med 2014; 371:434-46.
- Antoine C, Muller S, Cant A, Cavazzana-Calvo M, Veys P, Vossen J, et al. Long-term survival and transplantation of haemopoietic stem cells for immunodeficiencies: report of the European experience 1968-99. Lancet 2003; 361: 553-60.
- Mazzolari E, Forino C, Guerci S, Imberti L, Lanfranchi A, Porta F, et al. Long-term immune reconstitution and clinical outcome after stem cell transplantation for severe T-cell immunodeficiency. J Allergy Clin Immunol 2007; 120:892-9.
- Neven B, Leroy S, Decaluwe H, Le Deist F, Picard C, Moshous D, et al. Long-term outcome after hematopoietic stem cell transplantation of a single-center cohort of 90 patients with severe combined immunodeficiency. Blood 2009; 113:4114-24.
- Gennery AR, Cant AJ. Advances in hematopoietic stem cell transplantation for primary immunodeficiency. Immunol Allergy Clin North Am 2008; 28(2):439-56, x-xi. doi: 10. 1016/j.iac.2008.01.006.
- Norman M, David C, Wainstein B, Zeigler JB, Cohn R, Mitchell R, et al. Haematopoietic stem cell transplantation for primary immunodeficiency syndromes: A 5-year single-center experience. J Paediatr Child Health 2017; 53(10): 988–94.
- Laberko A, Gennery AR. Clinical considerations in the hematopoietic stem cell transplant management of primary immunodeficiencies. Expert Rev Clin Immunol 2018; 14(4): 297-306.
- Hladun R, Badell I, González M, Martínez AM, Sánchez de Toledo J, Olivé MT, et al. Survival analysis of hematopoietic stem cell transplantation in children with primary immune disorders. Anales de Pediatría 2015; 82(2):62-7. doi.org/10. 1016/j.anpede.2014.12.001.