Coexistence of Colon Perforation and Congenital Lipodystrophy
By Nihan Acar1, Turan Acar1, Beste Suataman1, Nese Ekinci2, Fatma Tatar1Affiliations
doi: 10.29271/jcpsp.2022.09.1222ABSTRACT
Lipodystrophy (LD) is an acquired or congenital rare condition consisting of hyperlipidaemia, glucose intolerance/ insulin resistance, and almost complete absence and storage of adipose tissue. Colon perforations can be observed in type 4 congenital LD. Here, we aimed to present a case of sigmoid colon perforation which developed in a young woman with the diagnosis of LD. Extensive purulent peritonitis, significant wall thickening, and oedema in the sigmoid colon were detected during surgical exploration. Anterior resection with end colostomy procedure was then performed.
Although bowel perforation has been theoretically reported to occur in LD, the presented case is the first adult patient in the literature. These individuals tend to develop colon perforation as a result of histological changes in their gastrointestinal tract. This situation should always be taken into consideration in order to avoid delay in diagnosis, especially in patients who present with abdominal pain and have a history of LD.
Key Words: Intestinal perforation, Congenital lipodystrophy, Peritonitis, Sigmoid colon.
INTRODUCTION
Lipodystrophy (LD) is an acquired or congenital rare condition consisting of hyperlipidaemia, glucose intolerance/insulin resistance, and an almost complete absence or disorder of the storage of adipose tissue. The congenital form is classified into four subtypes, each manifesting different clinical features, according to the four different biallelic mutations.1 Among these four types, type 4 is characterised by the generalised lipodystrophy, congenital myopathy with high- serum creatine kinase levels, gastrointestinal dysmotility, skeletal abnormalities, and cardiac arrhythmias.2
Here, we aimed to present a case and management of sigmoid colon perforation which developed in a young woman with the diagnosis of congenital LD, which is the second case in all the age groups and first in an adult patient in the literature.
CASE REPORT
A 20-year female with type 4 congenital LD was admitted to the emergency department after two days of severe abdominal pain and nausea. Diagnosis of type 4 congenital LD had been made after genetic testing at another medical centre.
She had a history of insulin-dependent diabetes, hypertriglyceridemia, and ventricular tachycardia which was controlled with an implantable cardioverter defibrillator (ICD), scoliosis, and chronic constipation. She had been using propranolol, verapamil, flecainide, fenofibrate, gliclazide, and both short and long-acting insulin.
The patient was in good general condition, and initial vital signs revealed a heart rate of 110 beats/minute, normal blood pressure (120/70 mmHg), and subfebrile temperature (37.8°C). She had a pseudo-acromegaloid appearance, generalised loss of subcutaneous fat, and scoliosis. Physical examination displayed abdominal distension, diffuse tenderness, rebound in the left lower quadrant, and a normal digital rectal examination. Laboratory tests were unremarkable except for slightly elevated C-reactive protein (6.34 mg/dL). The abdominal X-rays and ultrasound did not give any clues for a preliminary diagnosis. Therefore, an abdominal computed tomography was performed which showed extra-luminal gas bubbles, intra-abdominal free fluid, diffuse wall thickening in the rectosigmoid and sigmoid colon, and pericolic stranding around the rectosigmoid colon (Figure 1). An emergency operation was planned due to the gastrointestinal perforation. Extensive purulent peritonitis, significant wall thickening, and oedema in the sigmoid colon were detected during the exploration. It was considered as perforated diverticulitis. Anterior resection with end colostomy was then performed. The patient was discharged uneventfully on the fifth postoperative day. Histopathological examination of the specimen revealed fibrosis, significant muscular hypertrophy, and loss of circular and longitudinal layer organisation of the muscularis propria. In addition, heavy granulation tissue was found on the serosal aspect of the colon but no signs of diverticula could be found (Figure 2a, 2b).
The patient had been followed up for a year without any complications and she did not prefer a second operation for the colostomy closure.
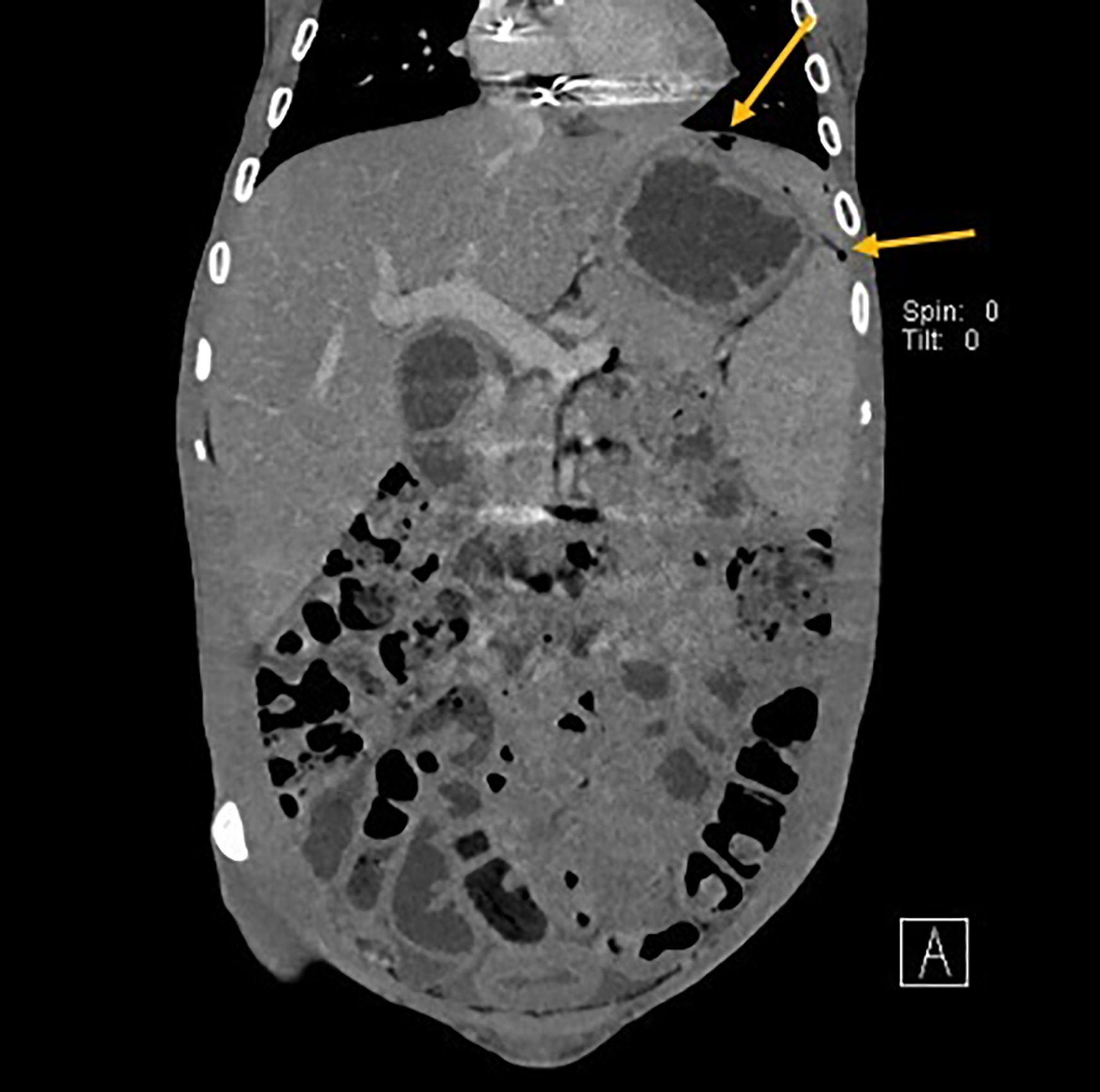 Figure 1: Abdominal computed tomography image in the coronal plane indicating gastrointestinal perforation (Arrows: Sub-diaphragmatic free air).
Figure 1: Abdominal computed tomography image in the coronal plane indicating gastrointestinal perforation (Arrows: Sub-diaphragmatic free air).
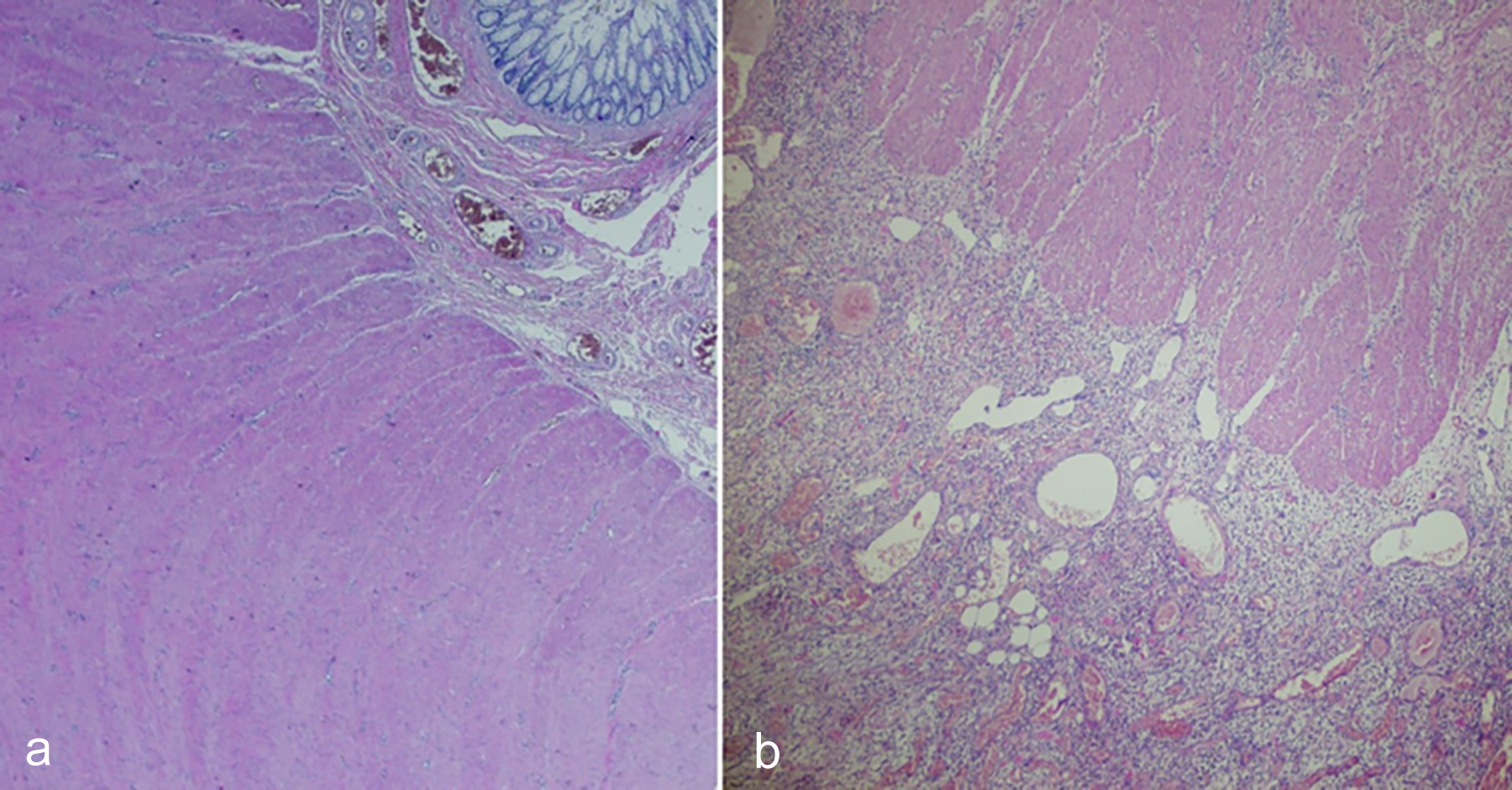 Figure 2: (a) Fibrosis and loss of circular and longitudinal layer organisation of the muscularis propria (H&E, ×20); (b) Granulation tissue on the serosal surface (H&E, ×40).
Figure 2: (a) Fibrosis and loss of circular and longitudinal layer organisation of the muscularis propria (H&E, ×20); (b) Granulation tissue on the serosal surface (H&E, ×40).
DISCUSSION
Munoz et al. were the first to describe a bowel perforation in a paediatric patient with type 4 congenital generalised LD in 2019.3 Since then, to the best of our knowledge, this is the second case in all the age groups and the first in adult patients in the literature.
Similar to the case of Munoz et al., this patient also had significant muscular hypertrophy and thickening in the colon wall.3 Muscular hypertrophy in these patients, just as the formation of colonic diverticula, may occur due to the gastrointestinal dysmotility and increased intracolonic pressure. Rajab et al. reported that smooth muscle hypertrophy, which was common in patients with type 4 congenital LD, was responsible for gastrointestinal dysmotility, dysphagia, ileus and infantile hypertrophic pyloric stenosis.4
In terms of surgical management, our approach to this case was similar to the treatment of perforated diverticulitis with purulent peritonitis. Although there have been many publications debating the ideal treatment choice for complicated diverticulitis, primary anastomosis with or without a diverting stoma is shown to be feasible as long as the patient’s condition is suitable and there is healthy colonic tissue for anastomosis.5 In our case, anastomosis was not preferred since the patient had peritonitis, multiple comorbidities, and the colon wall of the proximal end was edematous and extremely thick.
In their study reviewing the mortality of 502 patients with congenital LD, Gupta et al. reported that 33 patients died, and among the patients with a known cause of death, only one had peritonitis.6 Although our patient had high-risk cardiac comorbidities, mortality did not occur owing to the timely intervention before sepsis developed.
In conclusion, it is important for the general surgeons to be aware of the fact that LD individuals tend to develop colon perforation as a result of histological changes in their gastrointestinal tract. This situation should always be taken into consideration in order to avoid delay in the diagnosis, especially in patients who present with the abdominal pain and have a history of LD. In addition, we observed that there is limited clinical experience on this subject, and we believe our experience will guide the clinicians.
ACKNOWLEDGEMENT:
The authors thank all the general surgery staff for their cooperation.
PATIENT’S CONSENT:
Written consent was obtained from both the patient and her parents.
COMPETING INTEREST:
The authors declared no competing interest.
AUTHORS’ CONTRIBUTION:
NA: Concept, design, resource, data collection, analysis and/or interpretation, literature search, writing manuscript, and final approval.
TA: Resource, data collection, literature search, writing manuscript, critical reviews, and final approval.
BS: Design, data collection, and final approval.
NE: Resource, analysis and/or interpretation, critical reviews, and final approval.
FT: Analysis and/or interpretation, critical reviews, and final approval.
REFERENCES
- Guneş N, Kutlu T, Tekant GT, Eroglu AG, Ustundag NC, Ozturk B, et al. Congenital generalized lipodystrophy: The evaluation of clinical follow-up findings in a series of five patients with type 1 and two patients with type 4. Eur J Med Genet 2020; 63(4):103819. doi: 10.1016/j.ejmg.2019. 103819.
- Akinci G, Topaloglu H, Akinci B, Onay H, Karadeniz C, Ergul Y, et al. Spectrum of clinical manifestations in two young Turkish patients with congenital generalised lipodystrophy type 4. Eur J Med Genet 2016; 59(6-7):320-4. doi: 10.1016/j.ejmg.2016.05.001.
- Muñoz A, Radulescu A, Baerg J, Mendez Y, Khan FA. Bowel perforation in a pediatric patient with congenital generalised lipodystrophy type 4. JPS Case Reports 2019; 48:101257. doi: 10.1016/j.epsc.2019.101257.
- Rajab A, Straub V, McCann LJ, Seelow D, Varon R, Barresi R, et al. Fatal cardiac arrhythmia and long-QT syndrome in a new form of congenital generalised lipodystrophy with muscle rippling (CGL4) due to PTRF-CAVIN mutations. PLoS Genet 2010; 6(3):1000874. doi: 10.1371/journal.pgen. 1000874.
- Halim H, Askari A, Nunn R, Hollingshead J. Primary resection anastomosis versus Hartmann's procedure in Hinchey III and IV diverticulitis. World J Emerg Surg 2019; 14:32. doi: 10.1186/s13017-019-0251-4.
- Gupta N, Asi N, Farah W, Almasri J, Barrionuevo P, Alsawas M, et al. Clinical features and management of non-HIV-related lipodystrophy in children: A systematic review. J Clin Endocrinol Metab 2017; 102(2):363-74. doi: 10.1210/ jc.2016-2271.