Biotin-responsive Multiple Carboxylase Deficiency (MCD)
By Hafsa Majid, Sibtain Ahmed, Siraj Muneer, Ruqaiyyah Hamid, Lena Jafri, Aysha Habib KhanAffiliations
doi: 10.29271/jcpsp.2022.06.823ABSTRACT
This study aimed to determine the clinical spectrum and biochemical findings on urine organic acids (UOA) in Biotin-responsive multiple carboxylase deficiency (MCD) patients presenting to the biochemical genetics laboratory (BGL). Patients reported as MCD, from January 2013-December 2020 were included. The UOA was analysed by gas chromatography mass spectrometer. Demographic, clinical, and biochemical details were extracted from the BGL history form. Two hundred and two patients were reported to have biotin responsive MCD with 111(55%) males and a median (Q3-Q1) age of 7 months (13-4). Of these 71.7% (n=145) patients presented in infantile period. Parental consanguinity was observed in 80% (n=161) and another 32.6% (n=66) cases grandparents were cousins. The main presenting features were seizures, developmental delay, and lethargy. The common peaks were determined on UOA 3OHIVA, MC and MCC. MCD is not rare in Pakistani population; it is recommended to include this disorder in newborn screening programs.
Key Words: Biotin responsive multiple carboxylase deficiency, Organic acids, Amino acids, Pakistan, Inborn errors of metabolism.
Biotin is a cofactor required for the optimal activity of four biotin responsive carboxylase enzymes; propionic CoA Carboxylase and Methylcrotonyl CoA carboxylase involved in branched-chain amino acid metabolism; acetyl CoA carboxylase involved in Kreb’s cycle and pyruvate CoA Carboxylase involved in ketones metabolism.1 The bioactive biotin is free biotin, which is derived from two major sources; dietary sources including egg yolks, legumes, nuts, liver, sweet potatoes, mushrooms, bananas, broccoli, etc. The second major source is recycling of the available biocytin or biotinyl-peptides, cleaved from the biotin responsive carboxylase enzymes by the action of biotinidase.1 This cycle involves two major enzymes biotinidase and holocarboxylase synthase. The deficiency of any of the two enzymes leads to the deficient free biotin pool and in turn, decreased activity of the four biotin responsive multiple carboxylases, termed as biotin responsive multiple carboxylase deficiency (MCD). They are both autosomal recessive diseases, and assumed to have a higher prevalence in countries with higher consanguineous marriages rate, as seen in Pakistan. A wide spectrum of non-specific symptoms, including feeding difficulty, breathing difficulties, lethargy, seizures, skin rash, alopecia, and developmental delay are seen.2
The patients with the biotin responsive MCD are treated by prescribing high dose biotin and it is important to diagnose them to prescribe the amount needed. The Biotin-responsive MCD is diagnosed on the basis of excretion of a characteristic urine organic acid (UOA) pattern on gas chromatography-mass spectrometry (GC-MS).
The exact burden of biotin responsive MCD in Pakistan is unknown, only a few case reports have been published on this disease.3 Aim of this study was to determine the frequency of biotin responsive MCD in patients tested for UOA and determine the presenting pattern of these patients.
A descriptive, cross-sectional study was performed at the biochemical genetics laboratory (BGL), Section of Chemical Pathology, Department of Pathology and Laboratory Medicine, The Aga Khan University Hospital (AKU). The approval was taken from the institutes ethical review committee (ID# 2021-6008-17335). The UOA analysed at BGL from January 2013 to December 2020 was extracted. Only patients diagnosed with Biotin responsive MCD were included. While patients with peaks of valproate on chromatograms or clinical details forms mentioning valproate therapy, were excluded from the study. Clinical and biochemical history was collected from the test requisition forms of UOA. Details related to presenting pattern including age at the time of presentation, signs, symptoms, family history, and current status of the patients diagnosed with MCD were collected.
Qualitative evaluations of organic acids in urine were performed on agilent GCMS system (agilent technologies, US) using ethyl acetate, derivatisation by bis-(trimethylsilyl) acetamide compound and internal standard 3,3 dimethyl Glutaric acid (DMG). The MCD was diagnosed based on the presence of 3-hydroxyisovalerate (3OHIVA), 3-hydroxypropionate (3OHPA), 3-methylcrotonylglycine (MCC), Tiglylglycine (Tig), and methyl citrate (MC) peaks on UOA. Qualitative evaluation of the aforementioned peaks was done against the internal standard (3, 3 dimethyl glutaric acid) peak and interpreted as small, moderate, and large peaks.
The statistical analysis was performed using Microsoft Excel 2010. Histograms were plotted for normality assessment and medians and interquartile ranges (Q3-Q1) were reported for quantitative variables with skewed data. For qualitative variables frequency and percentages. The frequency of subjects presenting with MCD, their signs, symptoms, and family history were noted.
Out of 22,585 patients tested for UOA over 8 years’ period, 0.89% (n=202) patients had biotin responsive MCD with 111(55%) male, 91 (45%) females and median (Q3-Q1) age of 7 months (13-4). Of these 71.7% (n=145), and 4% (n=8) patients were <1years and >5 years old. Samples were received from all over Pakistan, with majority received from northern parts of the country 42% (n=85) from Khyber Pakhtunkhwa, 29% (n=59) from Punjab, 21% (n=42) from Sindh, 6% (n=12) from Islamabad, and 2% (n=4) from Afghanistan.
Clinical features of the patients were varied ranging from neurological to dermatological pathologies. Seizures were the most common clinical feature observed in 55.4% (n=112) patients, followed by developmental delay, lethargy 53% (n=107) and 50% (n=101) respectively (Figure 1). On UOA the most commonly found peaks were 3OHIVA, MC, MCC in 82% (n=164), 59.4% (n=120) and 56.4% (n=114) patients respectively.
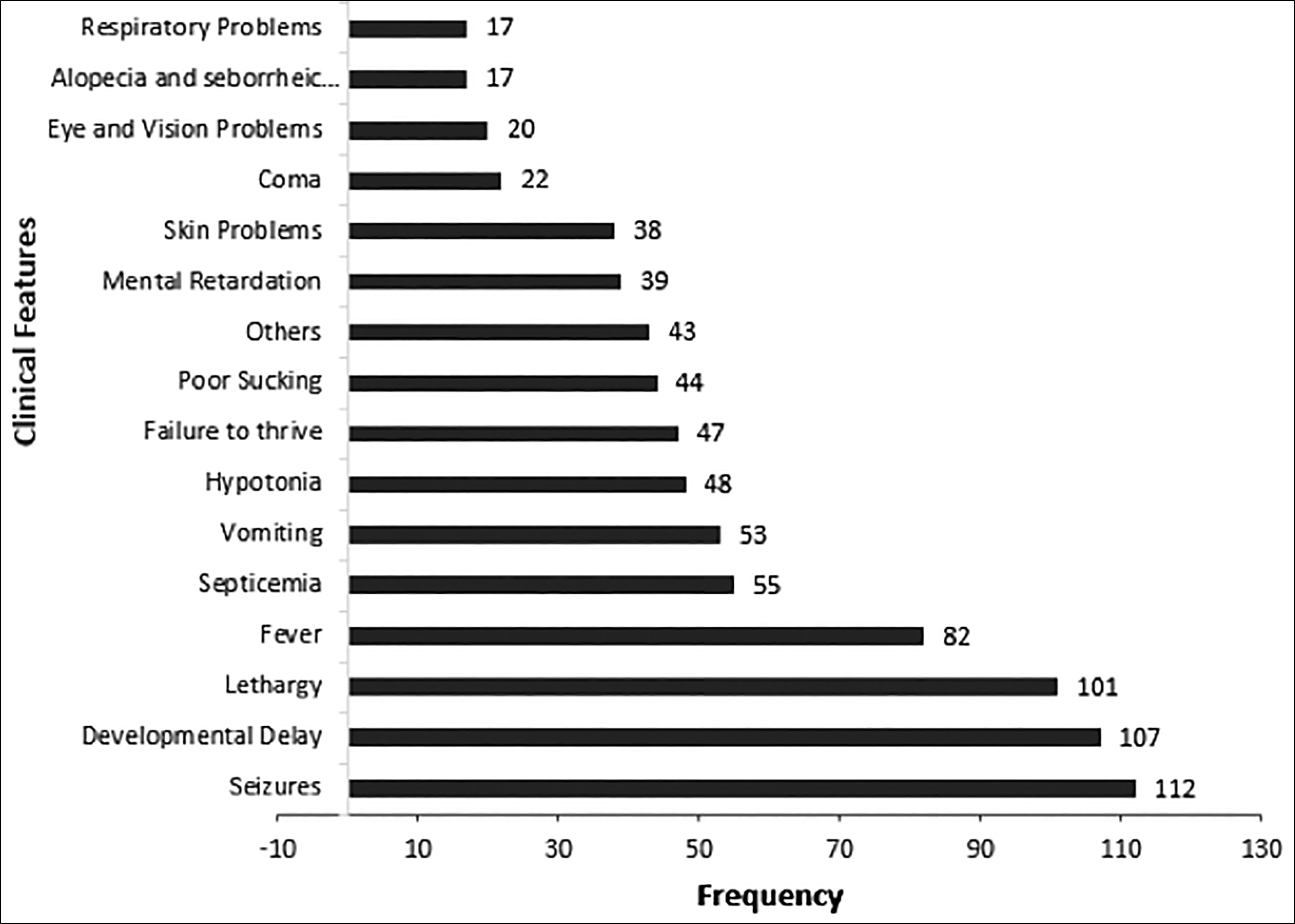 Figure 1: Frequency of clinical signs and symptoms of biotin responsive MCD patients (n=202).
Figure 1: Frequency of clinical signs and symptoms of biotin responsive MCD patients (n=202).
Metabolic acidosis was the most common biochemical findings. The median levels of the biochemical parameters, and organic acids are shown in Table I. Family history showed that parental consanguinity was observed in 80% (n=161) and another 32.6% (n=66) cases grandparents were cousins. Siblings of 8.5% (n=17) patients showed similar symptoms and 5.9% (n=12) of these expired. Of the total patients diagnosed with MCD, the condition of 6.4% (n=13) patients was deteriorating and 2 had expired.
Table I: The median levels of Biochemical Parameters for the patients diagnosed with biotin responsive MCD.
|
Biochemical Parameters |
Median (Q3-Q1) |
|
Routine Metabolic Parameters |
|
|
Blood pH (reference range 7.35-7.45) |
7.39 (7.43-7.2) |
|
Serum Sodium (reference range 135-145 mmol/L) |
139 (141-136) |
|
Serum Potassium (reference range 3.5-5.1 mmol/L) |
4.1 (4.5-3.5) |
|
Serum Chloride (reference range 98-107 mmol/L) |
107 (111-103) |
|
Total Bilirubin (reference range <1.2 mg/dl) |
0.36 (0.6-0.2) |
|
Serum Alanine Aminotransferase (Male <45 IU/L, female <35 IU/L) |
28.5 (46-19.2) |
|
Serum Alkaline phosphatase (reference range 54-369 IU/L) |
248 (356-166) |
|
Plasma Glucose (random) (reference range 80-160 mg/dl) |
110 (171-83) |
|
Urine Organic Acids |
|
|
3 hydroxy Isovaleric acid |
315.9 (635.7-126.3) |
|
3 Methylcrotonylgycine |
61.8 (175-25) |
|
Methyl Citric |
24.2 (39.7-16) |
|
Lactic acid |
323.6 (1155.4-94.9) |
|
Where urine organic acids were measured as pseudounits. |
|
The Biotin-responsive MCD encompasses two disorders biotinidase and holocarboxylase synthase deficiency, both of which are autosomal recessive and treatable disorders.1 Out of these, the incidence of biotinidase deficiency reported in the literature is higher, from 1: 6,000 to 1: 60,000 births than Holocarboxylase synthase deficiency, 1: 200,000 births. If left untreated these patients will develop irreversible neurological deficits and may even die. In many of the developed countries newborn screening for biotinidase deficiency is performed, so these salvageable patients are identified early and can be saved. Lack of awareness of the physicians or pediatricians and limited availability of diagnostic tests remains the two main challenges in identifying patients with MCD. The findings of the present study showed that the burden of biotin responsive MCD is high in our population, and presents evidence that UOA can be a good test for identifying patients with biotin responsive MCD. Another point to highlight was that the age of presentation was late as compared to the reported literature from other parts of the world, i.e. infantile or childhood period. The reason for this is that Pakistan does not have a newborn screening program for these disorders, hence the late presentation of patients.4 These findings also build a strong case for establishing newborn screening services for biotinidase deficiency.
In the present study, the clinical features of neurological deficit, i.e. seizures, developmental delay, and lethargy were the most commonly present, while skin problems, such as hair loss, and rash were noted in only 13.8% (n=38) patients. Similar findings were reported by a study from Bangladesh reporting neurometabolic disorders, including BTD, which states that developmental delay, seizures, and hypotonia were common symptoms. The parental consanguinity was reported in 60.9% cases, which is lower than this study (80% of cases).5 Another audit from India on organic acidurias reported that biotin responsive multiple carboxylase deficiency was common (33.33%) and the common clinical presentation of these patients were neurodevelopmental issues (73.3%) and metabolic crisis (53.3%).6 In the present study, the authors observed neurological deficit, however, any significant metabolic crisis was not observed.
The study limitation was that the patients were not differentiated into biotinidase and holocarboxylase deficiencies due to enzymes testing and gene mutation testing couldn’t be done owing to the non-availability of these tests locally. Further longitudinal multicenter studies are needed with categorisation of the two disorders.
In conclusion, the presently reported findings point towards an alarmingly high frequency of the biotin responsive MCD in Pakistan. The age of presentation is late and many of the patients have developed symptoms of neurological deficit at the time of diagnosis. Considering the high prevalence and easy availability of treatment for this disorder, it is recommended to include this disorder in national newborn screening program.
COMPETING INTEREST:
The authors declared no competing interest.
AUTHORS’ CONTRIBUTION:
HM: Conceived the idea, oversight of the project, data collection, analysis, and manuscript writing.
SA, SM: Involved in data analysis and reviewed the manuscript.
RH: Involved in data collection analysis and writing the manuscript.
LJ, AHK: Critically reviewed the manuscript for intellectual content.
All authors approved the final version of the manuscript to be published.
REFERENCES
- Leon-Del-Rio A. Biotin in metabolism, gene expression, and human disease. J Inherit Metab Dis 2019; 42(4):647-54.
- Akgun A, Sen A, Onal H. Clinical, biochemical and genotypical characteristics in biotinidase deficiency. J Pediatr Endocrinol Metab 2021; 34(11):1425-33. doi: 10.1515/jpem-2021-0242.
- Afroze B, Wasay M. Diagnosis, treatment and follow-up in four children with biotinidase deficiency from Pakistan. J Coll Physicians Surg Pak 2013; 23(10):823-5.
- Maguolo A, Rodella G, Dianin A, Monge I, Messina M, Rigotti E, et al. Newborn screening for biotinidase deficiency. The experience of a regional center in Italy. Front Pediatr 2021; 9:661416. doi: 10.3389/fped.2021.661416.
- Kundu GK IR, Ahmed S. Clinico-metabolic profile of neurometabolic disorders of children in a tertiary care Hospital of Bangladesh. European J Clin Med 2021; 2(4):1-5.
- Sindgikar SP, Shenoy KD, Kamath N, Shenoy R. Audit of organic acidurias from a single centre: Clinical and metabolic profile at presentation with long term outcome. J Clin Diagn Res 2017; 11(9):SC11-SC4. doi: 10.7860/JCDR/ 2017/28793.10632.