Berardinelli-Seip Congenital Lipodystrophy – A Case Report and Review of Literature
By Atif Munir, Miqdad Haider, Aijaz zeeshan Khan ChacharAffiliations
doi: 10.29271/jcpsp.2022.06.817ABSTRACT
Berardinelli-Seip Congenital Lipodystrophy (BSCL), also known as congenital generalised lipodystrophy, is a genetic disorder where there is an absolute deficiency of adipose tissue. It affects the development of adipocytes and ultimately leads to an inability to store fat in adipocytes. It is extremely rare. Most of the cases reported are from Africa and North America. Only a handful of cases have been reported in the world. The aim of this case report is to highlight the significance of this rare metabolic disorder, which should be considered whilst managing young patients with severe insulin resistance. We present a case of a young Asian child with an increasing need for insulin for his diabetes. He was previously managed on the lines of type 1 diabetes mellitus and his insulin requirements kept on increasing. Diagnosis on the basis of genetic studies was not possible due to the non-availability of the test in Pakistan. BSCL is an infrequent condition leading to several cardiometabolic complications. Timely diagnosis can lead to better management and prevention of complications.
Keywords: Insulin resistance, Lipodystrophy, Acanthosis nigricans, Hypertriglyceridemia, Genetic disease.
INTRODUCTION
Berardinelli-Seip congenital lipodystrophy (BSCL) has an estimated prevalence of 1 in 10 million people worldwide.1 It is inherited in an autosomal recessive manner. It is caused by mutations in the AGPAT2 (1-Acylglycerol-3-Phosphate O-Acyltransferase 2), BSCL2 (Berardinelli-Seip Congenital Lipodystrophy 2), CAV1 (Caveolin-1), and CAVIN1 (Caveolae-associated protein 1) genes. Anyone of these four mutations can lead to a specific type of lipodystrophy. Subcutaneous fat is scarce in this condition. Fat is deposited in body organs, predominantly in the liver. The patient usually has a muscular appearance with acromegaloid features.
Non-stored lipids cause hypertriglyceridemia or deposit in other sites like the liver.2 Patients usually have less than 2% of total body fat.3 The paucity of adipose tissue results in a decrease in leptin levels, which increases appetite; hence proving a hurdle in making patient compliant with dietary restrictions for Diabetes. Fat deposition in the liver can result in cirrhosis. All this explains the severity of this disease, which can result in early mortality.
A study conducted by Lima et al.1 to assess the life expectancy and cause of death in these patients concluded that BSCL leads to premature death, cutting the patients’ lifespan by 30 or more years. The majority of these young patients died of liver disease or infection and the same study concluded that more multicentre studies are required as a collaborative approach to a better understanding of this rare disease, to comprehend the mechanisms that predispose to infections, as well as to assess whether new therapies in the armamentarium can alter the natural course of this disease.
Diagnosis is made on history, physical examination, and genetic testing is confirmatory. Treatment is by restricting fat intake, diabetes control, and leptin.4 We report a case of this rare condition who presented to us with uncontrolled diabetes which was being treated on the lines of type 1 diabetes mellitus.
CASE REPORT
A 13-year boy was referred to our centre for uncontrolled diabetes. He was labelled as having Type 1 Diabetes, which was diagnosed a year ago and continued to have suboptimal glycaemic control despite good concordance with gradually escalating doses of insulin. There was no history of any episode of diabetic ketoacidosis (DKA). The boy had learning difficulties.
On examination, he had a muscular appearance with a typical facial appearance suggestive of lack of adipose tissue (Figure 1A). There was an obvious generalised lack of adipose tissue (Figure 1B) with thin but prominent muscular arms and legs with a protuberant abdomen. There was hepatomegaly. Widespread generalised acanthosis nigricans was present, most marked in the axilla.
Hemoglobin A1c (HbA1c) was 12%. Liver function tests were deranged with mild elevations of alanine aminotransferase (ALT) and aspartate aminotransferase (AST). The lipid profile was unremarkable. Abdominal ultrasound showed a fatty liver with early cirrhotic changes. The echocardiogram was unremarkable.
BSCL was suspected on clinical grounds. Genetic testing was not available for confirmation. Metformin was added to overcome insulin resistance, which resulted in optimisation of glycaemic control along with a total dose of 180 units of insulin daily. Leptin was not offered due to non-availability.
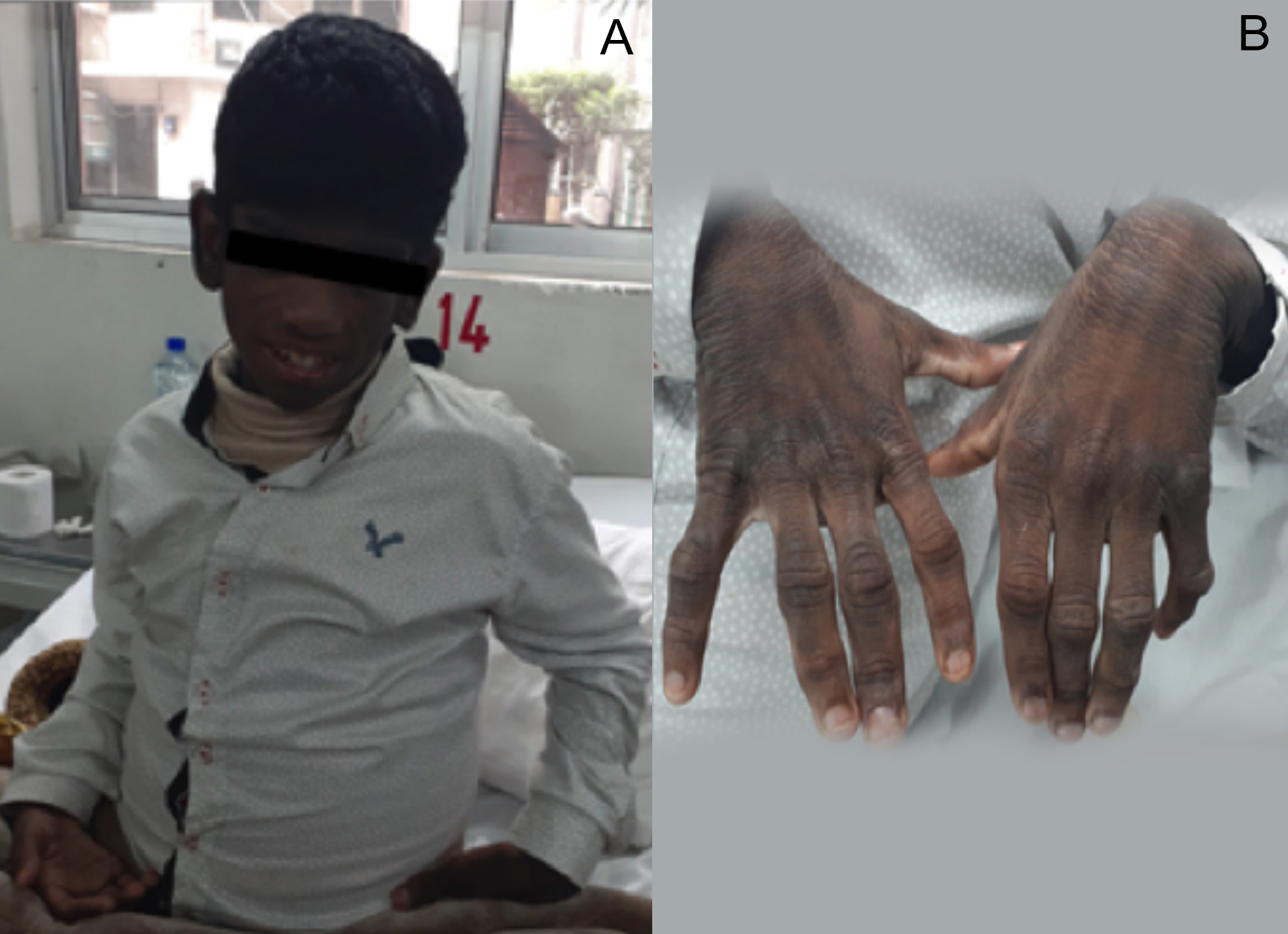 Figure 1: (A) Typical facial appearance of the patient. (B) Lack of adipose tissue on the back of hands.
Figure 1: (A) Typical facial appearance of the patient. (B) Lack of adipose tissue on the back of hands.
DISCUSSION
BSCL is an autosomal recessive condition, where there is a lack of fat tissue. The severity of the disease depends on the amount of fat loss and the resulting complications.5 Berardinelli from Brazil and Seip from Norway were the first ones to report cases of this disease in the 1950s.6 Between 300 and 500 people with the condition have been described in the medical literature and as per the best of our knowledge and literature review, only one case series with 5 cases have been reported in Pakistan to date.7
As per studies done on this disease, the probable cause of this disease is a defect in genes responsible for fat synthesis. The deficiency of fat results in a lack of a hormone called leptin, which is responsible for fat and glucose metabolism. People with BSCL generally have a peculiar appearance, where there is a dominance of body muscles along with a scarcity of adipose tissue.8 Usually clinical features and associated complications appear since early childhood. These include severe insulin resistance leading to Diabetes Mellitus (onset at around 15 years of age), xanthomas and pancreatitis secondary to hypertriglyceridemia, hepatomegaly, and liver failure secondary to hepatic steatosis, and hypertrophic cardiomyopathy (in 20% of cases). High levels of circulating insulin can lead to acanthosis nigricans.9
Diagnosis of this disorder is a challenging task. For a definite diagnosis, genetic studies must be carried out. Management includes dietary restrictions especially fat intake need to be reduced and proper education of parents in order to prevent undesirable stress and psychological sequelae in children affected with BSCL.10 Restriction of total fat intake to 20%-30% of total dietary energy helps to maintain normal serum triglyceride concentration. Leptin therapy for the treatment of hypertriglyceridemia and diabetes should be considered in these patients. Diabetes mellitus is managed as childhood-onset diabetes mellitus.
Loss of adipose tissue in this condition is not reversible; however, cosmetic procedures can help in improving physical features. Treatment of complications is the only option to improve quality of life. Metreleptin was approved by the food and drug administration authority (FDA) in the USA in the year 2014 to treat leptin deficiency.10
BSCL is inherited in an autosomal recessive manner. Carrier testing should be encouraged for high-risk relatives and pre-natal testing needs to be made compulsory for pregnancies at increased risk if the pathogenic variants are detected in the family.
There is a need for increasing awareness about this condition among physicians so that an early diagnosis along with a multidisciplinary treatment approach is adopted in order to prevent complications. Genetic counseling should be an integral part of the management of this disease.
PATIENT’S CONSENT:
Informed consent was obtained, from the patient’s guardian to publish this case.
COMPETING INTEREST:
The authors declared no competing interest.
AUTHORS’ CONTRIBUTION:
AM: Clinical care diagnostic workup, and write up.
MH: Write up, discussion, and literature search.
AZKC: Literature search, proofreading.
All authors approved the final version of the manuscript to be published.
REFERENCES
- Lima JG, Nobrega LH, Lima NN, dos Santos MC, Silva PH, Baracho MD, et al. Causes of death in patients with Berardinelli-Seip congenital generalised lipodystrophy. Plos One 2018; 13(6). doi.org/10.1371/journal.pone.0199052.
- Debray FG, Baguette C, Colinet S, Van Maldergem L, Verellen-Dumouin C. Early infantile cardiomyopathy and liver disease: A multisystemic disorder caused by congenital lipodystrophy. Mol Genet Metab 2013; 109(2):227-9. doi.org/10.1016/j.ymgme.2013.04.011.
- Lima JG, Nobrega LH, de Lima NN, do Nascimento Santos MG, Baracho MF, Jeronimo SM. Clinical and laboratory data of a large series of patients with congenital generalised lipodystrophy. Diabetol Metab Syndr 2016; 8(1):23. doi.org/10.1186/s13098-016-0140-x.
- Zhou H, Li J, Su H, Li J, Young ME, CHEN W. Berardinelli-seip congenital lipodystrophy 2 is essential in regulating cardiac substrate metabolism and function. Circulation 2019; 140(Suppl_1):A11930. doi/abs/10.1161/circ.140.suppl_1. 11930.
- de Medeiros JL, Bezerra BC, de Araújo TA, Sarmento AS, de Azevedo Medeiros LB, Gualdi LP, et al. Impairment of respiratory muscle strength in Berardinelli-Seip congenital lipodystrophy subjects. Respir Res 2018; 19(1):173. doi.org/10.1186/s12931-018-0879-8.
- de Azevedo Medeiros LB, Dantas VK, Sarmento AS, Agnez-Lima LF, Meireles AL, Nobre TT, et al. High prevalence of Berardinelli-Seip congenital lipodystrophy in Rio Grande do Norte State, Northeast Brazil. Diabetol Metab Syndr 2017; 9(1):80. doi.org/10.1186/s13098-017-0280-7.
- Cheema HA, Malik HS, Waheed N, Mushtaq I, Fayyaz Z, Anjum MN. Berardinelli-seip congenital generalised lipodystrophy. J Coll Phys Surg Pak 2018; 28(5):406-8. doi: 10.29271/jcpsp.2018.05.406.
- Lima JG, Nobrega LH, Lima NN, dos Santos MC, Maria de Fatima PB, Bandeira F, et al. Bone density in patients with berardinelli-seip congenital lipodystrophy is higher in trabecular sites and in type 2 patients. J Clin Densitometry 2018; 21(1):61-7. doi.org/10.1016/j.jocd.2016.10.002.
- Cândido Dantas VK, da Silva Soares J, de Azevedo Medeiros LB, Sarmento AS, de Azevedo Medeiros B, Craveiro Sarmento AS, et al. Nurses' knowledge about Berardinelli-seip congenital lipodystrophy. Plos One 2018; 13(6):0197784. doi.org/10.1371/journal.pone.0197784.
- Joshi R, Sharma S. Berardinelliseip congenital lipodystrophy syndrome: 10 year follow-up. Indian Pediatr 2019; 56(9):877-8. doi.org/10.1007/s13312-019-1617-0.