A Delayed Presentation of Arginase Deficiency Presenting with Status Epilepticus
By Asburce Olgac1, Eren Yildiz2, Arzu Yilmaz3, Cigdem Seher Kasapkara1, Serdar Ceylaner4, Bulent Alioglu5Affiliations
doi: 10.29271/jcpsp.2022.12.1629ABSTRACT
Arginase 1(ARG1) deficiency is a rare disorder of the urea cycle. The presentation is usually late, leading to loss of intellectual milestones, spasticity and liver involvement. Hyperammonemic crises are rarely encountered. We herein present a case of a 16-year immigrant girl of Syrian origin who was evaluated for acute onset of fever, vomiting, and seizures. Laboratory analyses showed slightly elevated lactate, creatine kinase, and coagulation parameters. Ammonium levels were also moderately increased. On 5th day of admission, she went into an encephalopathic state. Blood amino acid analysis showed highly elevated arginine levels. An increased level of orotic acid was found in urine organic acid analysis. Molecular genetic analysis of ARG1 gene showed a novel homozygous mutation.
Although the presentation of ARG1 deficiency is usually chronic in the majority of patients, an acute crisis of encephalopathy due to hyperammonemia may occur and delayed diagnosis may lead to irreversible neurological damage.
Key Words: Urea cycle disorder, Hyperammonemia, Argininemia, Encephalopathy.
INTRODUCTION
Arginase 1 (ARG 1) deficiency is a rare, autosomal recessively inherited urea cycle disorder (UCD). The incidence is reported to be between 1:350,000 and 1:1,000,000.1 In contrast to the majority of UCDs, in which hyperammonemia in the newborn period is the classical presentation, hyperammonemic crises are not usual. Some patients may be misdiagnosed as having cerebral palsy (CP) since the disease causes spasticity and progressive intellectual impairment.1,2 The underlying aetiology of neurological symptoms in ARG 1 deficiency is suggested to be the accumulation of arginine or guanidine compounds that cause neurotoxicity.2
We present an atypical case of ARG 1 deficiency due to a novel mutation presenting with encephalopathy.
CASE REPORT
A 16-year immigrant girl of Syrian origin was evaluated for acute onset of fever and vomiting. She was the 6th child of consanguineous parents. Vomiting and fever had started acutely and she was admitted to the intensive care unit (ICU) after generalised seizures, where midazolam and phenytoin infusion was initiated.
Physical examination showed growth retardation and deep tendon reflexes were increased. She could communicate and could perform simple tasks. Psychometric tests could not be performed due to language limitations. Initial laboratory analyses showed normal complete blood count and blood gas analysis. C-reactive protein was greatly increased (100 mg/dl, normal range [NR]:0-5). Lactate (2.8 mmol/L, NR: <2 mmol/L), creatine kinase (314 IU/L, NR: 41-277), and ammonium levels (155 mmol/L, NR: 19-30) were slightly elevated. Other biochemical makers, cerebrospinal fluid (CSF) microscopy, culture, and biochemical analyses were normal. Electroencephalography showed generalised spike and slow wave activity. Antibiotics due to suspicion of infection and antiepileptic treatment including 0.1 mg/kg/h midazolam infusion, phenytoin and carbamazepine were initiated.
Due to ongoing seizures, ammonium level was re-checked and was found to be increased (423 mmol/L) and ammonium reducing therapy including sodium phenylacetate and sodium benzoate infusion were administered. Carglumic acid, 100 mg/d, L-carnitine, 100 mg/kg/d and biotin, 10 mg/d were started enterally. Hydroxycobalamine, 1 mg/d, intramuscularly was also administered. Peritoneal dialysis was performed due to refractory hyperammonemia (660 mmol/L). After normalisation of ammonium levels, peritoneal dialysis was discontinued and sodium benzoate, 250 mg/kg/d, was initiated via nasogastric tube.
Cranial MRI showed symmetric cortical swelling, edematous signal changes and restricted diffusion at bilateral frontal, insular, and temporal lobes (Figure 1).
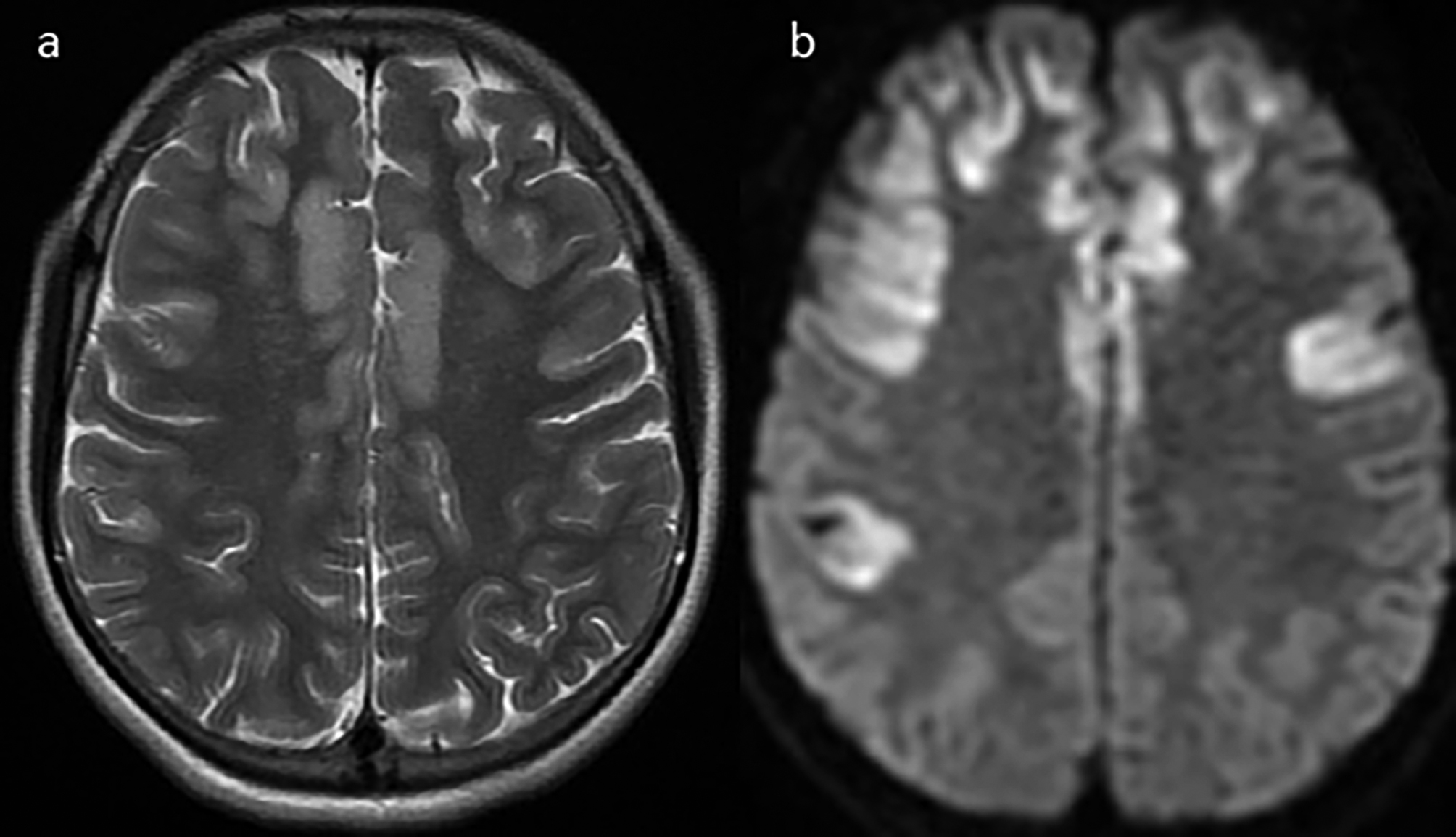 Figure 1(a): Axial plane T2-weighted brain MRI shows symmetric cortical swelling and edematous signal changes in bilateral frontal, insular and temporal lobes. (b): Axial plane of the diffusion-weighted MRI shows restricted diffusion in bilateral frontal, insular and temporal lobes.
Figure 1(a): Axial plane T2-weighted brain MRI shows symmetric cortical swelling and edematous signal changes in bilateral frontal, insular and temporal lobes. (b): Axial plane of the diffusion-weighted MRI shows restricted diffusion in bilateral frontal, insular and temporal lobes.
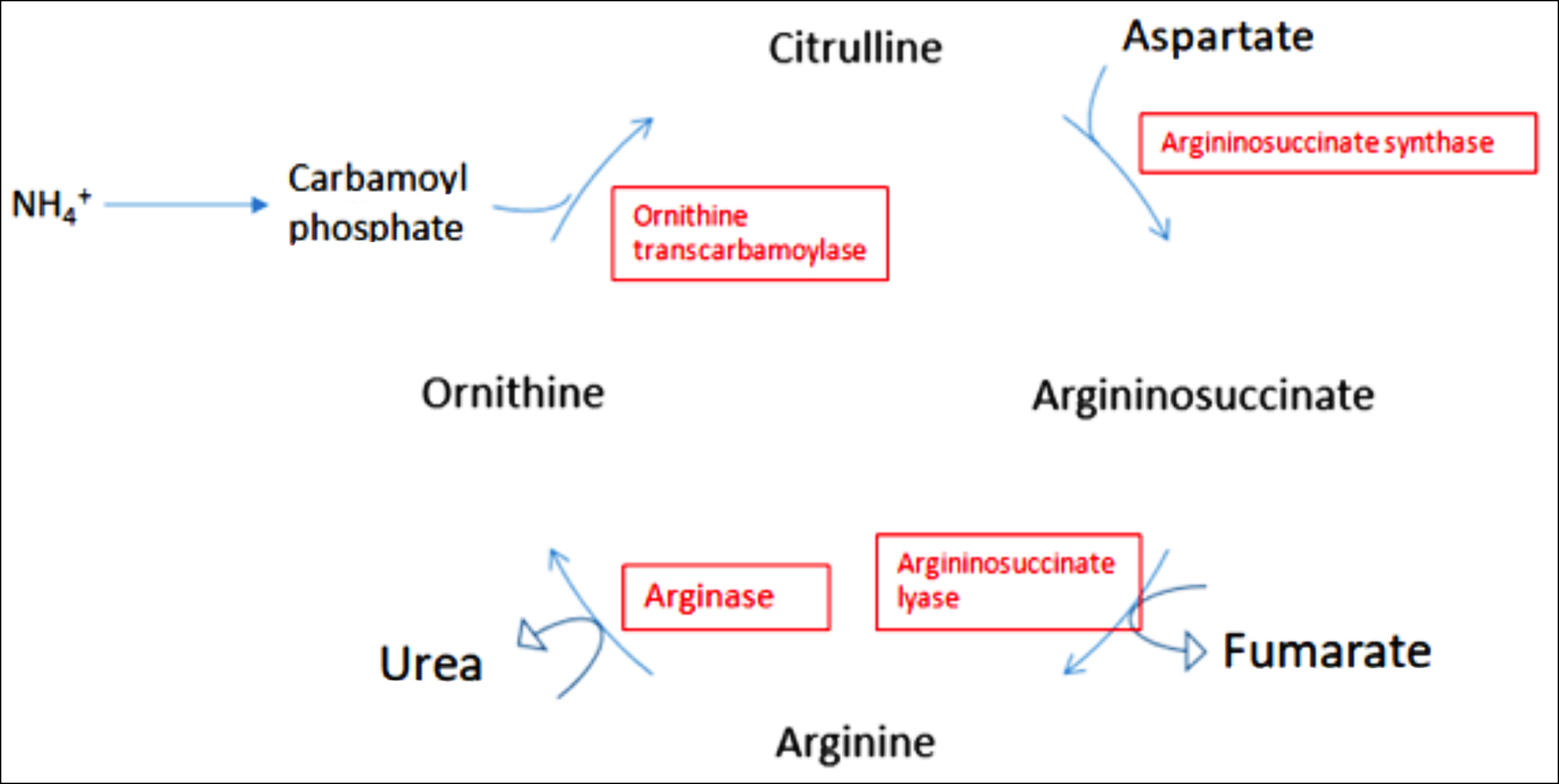 Figure 2: A schematic presentation of the urea cycle.
Figure 2: A schematic presentation of the urea cycle.
Metabolic tests were performed. Blood amino acid analysis by using high-performance liquid chromatography (HPLC) showed arginine to be 319.7 mmol/L (NR: 45-150). Urine organic acid analysis showed increased levels of orotic acid (20.62 mmol/mol Cr, NR: 0-1.9) in urine.
Her neurological condition gradually improved with dietary protein restriction of 1 g/kg/day and arginine levels were normalised (74.64 mmol/L, NR: 45-150). Unfortunately, she has still tetraparesis and lack of speech ability.
Molecular studies of the ARG1 gene showed a novel homozygous mutation in ENST00000356962.2:c.58-3C>G in intron 1. Human Splicing Finder 3.0 and Mutation Taster software predicted it as a pathogenic mutation. Parental DNA could not be analysed for this mutation due to loss of follow-up.
DISCUSSION
The urea cycle consists of five consecutive enzymatic reactions distributed between the mitochondria and cytosol (Figure 2).1-3 ARG1 enzyme is encoded by the ARG1 gene located on chromosome 6q23, encompassing a 15 kb region and comprising 8 exons.4 Its deficiency leads to the accumulation of arginine, which is a precursor for the synthesis of nitric oxide and guanidino compounds. ARG 1 deficiency differs from other UCDs in that patients are less prone to hyperammonemia.1-3
It typically manifests in childhood with mild to moderate hyperammonemia and neurological dysfunction including developmental delay, progressive spastic paraparesis (especially of the lower extremities), epileptic seizures, and occasionally ataxia and dystonia. ARG 1 deficiency may be misdiagnosed as CP. Hepatic pathology may also be present.1-3
Diagnosis may be made by using tandem mass spectrometry (MS/MS) or HPLC by determining elevated arginine levels, the biochemical hallmark of ARG 1 deficiency.4 Diagnosing the disease early (especially in the neonatal period) will lead to early initiation of treatment that may prevent neurological damage to some extent.5
Neuroradiological findings include mild to severe cerebral and cerebellar atrophy, signal changes in the posterior putamen and insular cortex, corticospinal tract alterations on diffusion tensor imaging (DTI), global brain oedema, basal ganglia involvement followed by delayed myelination, and cystic lesions.6
The most commonly encountered defects in the ARG1 gene are heterogeneous missense mutations that disrupt with or interfere with the active sites required for the catalytic reaction.1,2 Jain-Ghai et al.7 suggested severe mutations to cause neonatal presentation, while the classic later-onset ARG 1 deficiency patients have at least one missense mutation, as in this patient.
Treatment of ARG 1 deficiency consists of protein restriction in diet to lower plasma arginine levels, which may prevent further neurological deterioration.8
Physicians, especially paediatricians, paediatric neurologists, physiotherapists, and health professionals involved in the care of CP should be aware of inborn errors of metabolism that may present as spastic diplegia. Progressive neurological signs and atypical presentations of arginase deficiency are the red flags in CP that underlying metabolic aetiology should be considered.9
We presented a case of ARG 1 deficiency with a non-classical presentation. We would like to emphasise the importance of metabolic tests for the diagnosis and treatment of UCDs, since delays can result in severe neurological damage. Early recognition of the disease may prevent the devastating effects of hyperammonemia and may affect disease outcomes positively.
PATIENTS’ CONSENT:
Informed consent is obtained from the patient to publish the data concerning this case.
COMPETING INTEREST:
The authors declared no competing interest.
AUTHORS’ CONTRIBUTION:
AO, EY, AY, CSK, SC, BA: Designed and drafted the manuscript and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
All the authors have approved the final version of the manuscript to be published.
REFERENCES
- Matsufuji M, Takeshita E, Nakashima M, Watanabe Y, Fukui K, Hanai T, et al. Sodiumphenylbutyrate improved the clinical state in an adult patient with arginase 1 deficiency. Brain Dev 2020; 42(2):231-5. doi. 10.1016/j.braindev. 2019.09.002.
- Yuan YS, Garrett B, Andreas S, Colin DF. Arginase-1 deficiency. J Mol Med 2015; 93:1287-96. doi: 10.1007/ s00109-015-1354-3.
- Schlune S, Vom Dahl D, Haussinger D, Ensenauer R, Mayatepek E. Hyperargininemia due to arginase I deficiency: The original patients and their natural history and a review of the literature. Amino Acids 2015; 47(9):1751-62. doi: 10.1007/s00726-015-2032-z.
- Yokoi K, Nakajima Y, Yasui T, Yoshino M, Yoshikawa T, Kurahashi H, et al. Novel ARG1 variants identified in a patient with arginase 1 deficiency. Hum Genome Var 2021; 8(1):8. doi: 10.1038/s41439-021-00139-9.
- Merritt JL, Brody LL, Pino G, Rinaldo P. Newborn screening for proximal urea cycle disorders: Current evidence supporting recommendations for newborn screening. Mol Genet Metab 2018 124(2):109-13. doi: 10.1016/j. ymgme.2018.04.006.
- Carvalho DR, Brum JM, Speck-Martins CE, Ventura FD, Navarro MM, Coelho KE, et al. Clinical features and neurologic progression of hyperargininemia. Pediatr Neurol 2012; 46(6):369-74. doi: 10.1016/j.pediatrneurol. 2012.03. 016.
- Jain-Ghai S, Nagamani SC, Blaser S, Siriwardena K, Feigenbaum, A. Arginase I deficiency: Severe infantile presentation with hyperammonemia: More common than reported? Mol Genet Metab 2011; 104(1-2):107-11. doi: 10.1016/j.ymgme.2011.06.025.
- Baranello G, Alfei E, Martinelli D, Rizzetto M, Cazzaniga F, Dionisi-Vici C, et al. Hyperargininemia: 7-month follow-up under sodium benzoate therapy in an Italian child presenting progressive spastic paraparesis, cognitive decline, and novel mutation in ARG1 gene. Pediatr Neurol 2014; 51(3):430-3.
- Tsang JP, Poon WL, Luk HM, Fung CW, Ching CK, Mak CM, et al. Arginase deficiency with new phenotype and a novel mutation: Contemporary summary. Pediatr Neurol 2012; 47(4):263-9. doi:10.1016/j.pediatrneurol.2014.05.029.