Investigation of Hereditary Cancer Predisposition Genes of Patients with Colorectal Cancer: Single-centre Experience
By Neslihan Duzkale1, Ozlem Oz2, Tugba Taskin Turkmenoglu3, Kadir Cetinkaya4, Tulay Eren5, Suayip Yalcin6Affiliations
doi: 10.29271/jcpsp.2021.07.811ABSTRACT
Objective: To investigate the genetic causes of colorectal cancers (CRCs); and to determine the genotype-phenotype correlation.
Study Design: Descriptive study.
Place and Duration of Study: Department of Medical Genetics, Diskapi Yildirim Beyazit Training and Research, Hospital, Ankara, Turkey, between January 2018 and January 2020.
Methodology: 59 cancer susceptibility genes of 41 patients, included in the study and diagnosed with CRC, were examined using next generation sequencing (NGS) technique. Statistical analysis of the possible relationships among the mutation carrier status of the patients and the parameters of gender, age at diagnosis, and family cancer history, were performed.
Results: The mean age at diagnosis of all CRC patients was 48.7 years (range 28-74). Mutations in MLH1, MSH6, CHEK2, PMS2 and MUTYH genes were detected in 10 patients (24.4%). The mean age at diagnosis of CRC was 46.2 years in those who carried the mutation, while it was 49.5 years in those without. Carriers and non-mutation carriers, when compared in terms of age at diagnosis, gender, family cancer history, no significant difference was observed.
Conclusion: Genes that may cause susceptibility to cancer may play a role in the etiopathogenesis of the CRC. NGS-based multigene panels allow these genes to be detected in the patient and to identify an inherited cancer syndrome.
Key Words: Colorectal cancer, Lynch syndrome, Hereditary cancer, Gene, Next generation sequencing.
INTRODUCTION
Inherited cancer syndromes are a group of disorders caused by the inherited causative gene (s) that result in a higher risk of malignancy in the carrier individuals compared to the population risk. In individuals with this syndrome, findings such as many relatives with certain types of cancer, an earlier than expected cancer diagnosis age, or the development of two or more types of cancer in the same person can be observed.1-3
Colorectal cancers (CRCs) have an important place among hereditary cancers, and according to the latest statistics of the World Health Organisation, they rank third among all cancers in terms of incidence and second in terms of mortality.1 The incidence rates of this type of cancer vary greatly in many geographical regions and ethnic groups.2
Approximately 5% of the cases are associated with hereditary (inherited) mutations in well-characterised and highly penetrating cancer susceptibility genes. Genetic instability caused by these mutations triggers CRC development.3 The most common hereditary CRCs; hereditary non-polyposis colorectal cancer (HNPCC), familial adenomatosis polyposis (FAP), MUTYH associated polyposis (MAP), Cowden/PTEN hamartoma syndrome and other subtypes of this group. In general, HNPCC is responsible for about 2-4% of CRC cases and FAP for 1%. Less than 1% of cases are caused by MAP, hamartomatous polyposis and hyperplastic polyposis syndromes.4 Major susceptibility genes that cause HNPCC, also known as DNA Mismatch Repair (MMR) genes; While MLH1, MSH2, MSH6 and PMS2 are the gene responsible for FAP is APC. There is evidence that a series of complexities caused by mutations in the pathways in which these genes play a role accelerate the carcinogenesis process of these individuals.5
Recent advances in genetic testing technologies have accelerated the molecular diagnosis of hereditary cancer syndromes and the discovery of new susceptibility genes that cause CRC.
The aim of this study was to investigate the general demographic and clinicopathological features of the patients with CRC and the genetic causes that may have a role in the etiopathogenesis and pathophysiology of the disease.
METHODOLOGY
This observational study was conducted at the Department of Medical Genetics, Diskapi Yildirim Beyazit Training and Research, Hospital, Ankara, Turkey, between January 2018 and January 2020. Forty-one patients older than 18 years of age and diagnosed with CRC were included in the study. The gene analysis results, general demographic characteristics, clinical and pathological details, and family cancer history of these patients were obtained retrospectively from the patient files. In the study, germline variants of 59 hereditary cancer-associated genes in 41 patients were investigated. This study was conducted by considering ethical responsibilities, according to the World Medical Association and the Declaration of Helsinki; and the study protocol was approved by local Ethics Committee.
Genetic analysis of the patients was performed with DNA materials obtained from peripheral venous blood samples by next generation sequencing (NGS) method. This analysis was made using the Qiagen large hereditary cancer panel (Qiagen, Hilden, Germany) kit and on the platform of the Illumina MiSeq system (Illumina Inc., San Diego, CA, USA). Researched genes were listed as: AIP, APC, ATM, ATR, AXIN2, BAP1, BARD1, BLM, BMPR1A, BRCA1, BRCA2, BRIP1, BUB1B, CDH1, CDK4, CDKN2A, CHEK2, CTNNA1, EPCAM, FAM175A, FANCC, FLCN, GALNT12, , GREM1, HOXB13, MET, MLH1, MRE11A, MSH2, MSH6, MUTYH, NBN, NTHL1, PALB2, PALLD, PIK3CA, PMS1, PMS2, POLD1, PRSS1, PTCH1, PTEN, RAD50, RAD51B, RAD51C, RAD51D1 , SDHB, SDHC, SDHD, SMAD4, SMARCA4, STK11, TP53, VHL, XRCC2.
In the analysis of the data obtained by the NGS method "Qiagen Clinical Insight (QCI™) Analyse software (QIAGEN, Hilden, Germany)", which is a commercial bioinformatics programme. In the study, the detected gene variants were classified, based on the criteria in the ACMG guideline.6 Among the classified germline causal gene variants, those located in the homopolymer and splice region, those with a reading depth and allele fraction below 30X, insertion and/or deletion type variants, and novel genomic changes were validated by the Sanger sequencing technique. The data obtained were evaluated using Mann-Whitney U-test, Fisher-Exact test and Independent-sample t-test statistical methods. Package for Social Sciences (SPSS v 15.0, SPSS Inc., Chicago, IL, USA) was used in all statistical analyses and the statistical significance level was accepted as p <0.05.
RESULTS
In total 41 patients included in the study, 22 were females (53.7%), and 19 were males (46.3%), and the mean age at diagnosis was 48.7 (range 28-74) years. The histopathological diagnosis of CRC in all patients was of the adenocarcinoma type. As a result of genetic analysis, disease-related germline pathogenic and likely pathogenic gene variants were detected in 10 (24.4%) patients (Table I). There was also no significant relationship between mutation carriage and gender. With the statistical analysis performed, no difference was observed in the age of diagnosis of the patients according to gender (p = 0.329).
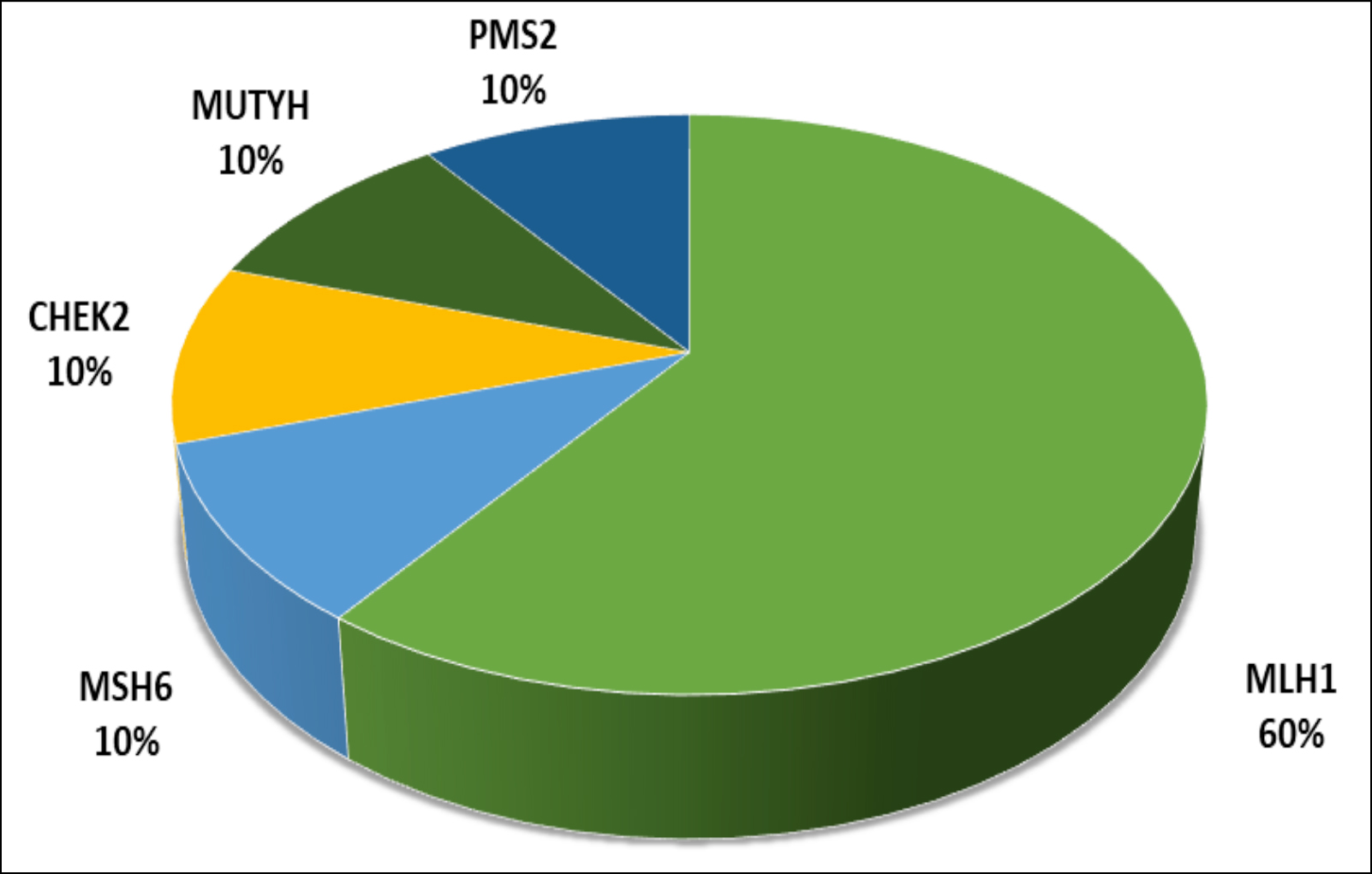 Figure 1: The detected causal genes and their distribution.
Figure 1: The detected causal genes and their distribution.
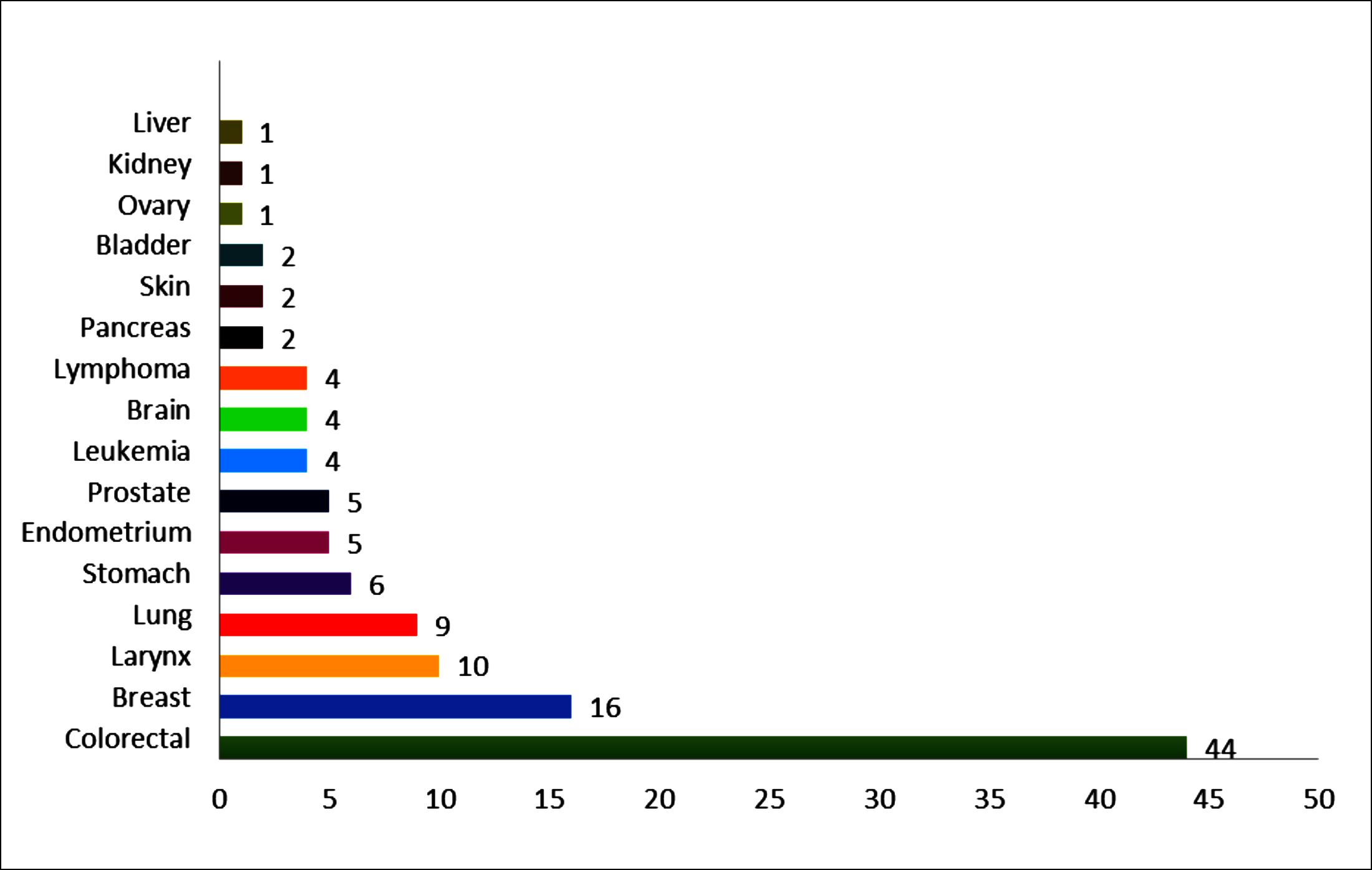 Figure 2: The number of relatives with cancer and primary sites/types of malignancy.
Figure 2: The number of relatives with cancer and primary sites/types of malignancy.
The horizontal axis shows the number of relatives diagnosed with cancer, and the vertical axis shows the types of cancer in these relatives.
In this study, the causative genes detected in 10 patients were determined as MLH1, MSH6, CHEK2, PMS2 and MUTYH (Figure 1). Although the genetic background of the remaining 31 patients (75.6%) could not be elucidated, variants of unknown clinical significance (VUS) were detected in the genes of 17 of these patients. VUS variants were most common in the ATM, APC, PALLD genes. The mean age at diagnosis of cancer in 10 patients, whose genetic reasons were clarified, was 46.2 (range 30-67) years, and the mean age at diagnosis of cancer was 49.5 (28-74) years in remaing patients. Statistical analysis showed that there was no significant relationship between age at the time of diagnosis and mutation carrying status (p = 0.481).
Table I: The list of all gene variants identified in this study, with patients’ demographic and clinical information included|
P ID/Sex |
Age/ Age at DX |
Gene |
Nucleotide Change |
AA Change |
Zyg |
Loc |
Func |
ACMG Scoring |
dbSNP |
Family history* |
|
P6/F |
70/67 |
MSH6 |
c.3226C>T |
p.Arg1076Cys |
Het |
Ex5 |
MS |
Pat |
rs63750617 |
1PR, 1LC, 2CRC, 1ST, BRA |
|
MUTYH |
c.884C>T |
p.Arg1076Cys |
Het |
Ex10 |
MS |
L.Pat |
rs374950566 |
|||
|
P7/F |
31/30 |
MLH1 |
c.1609C>T |
p.Gln537* |
Het |
Ex14 |
NS |
Pat |
rs63751277 |
4CRC |
|
P12/M |
38/35 |
MLH1 |
c.293G>C |
p.Gly98Ala |
Het |
Ex3 |
MS |
L.Pat |
- |
3CRC |
|
BARD1 |
c.1350T>G |
p.Asn450Lys |
Het |
Ex5 |
MS |
VUS |
rs876660753 |
|||
|
P17/F |
35/36 |
MLH1 |
c.677G>A |
p.Arg226Gln |
Het |
Ex8 |
MS |
Pat |
rs63751711 |
7CRC, 1PA |
|
P41/M |
36/35 |
MLH1 |
c.1896+2T>C |
- |
Het |
Int16 |
SE |
Pat |
rs267607869 |
- |
|
P21/M |
76/63 |
MUTYH |
c.884C>T |
p.Pro295Leu |
Hom |
Ex10 |
MS |
L.Pat |
rs374950566 |
1BRE, 1CRC |
|
ATM |
c.5870A>G |
p.Tyr1957Cys |
Het |
Ex39 |
MS |
VUS |
- |
|||
|
P24/M |
63/62 |
PMS2 |
c.690_691delGT |
p.Phe231Trpfs |
Het |
Ex6 |
FS |
Pat |
rs1064795447 |
1CRC |
|
P39/M |
50/40 |
MLH1 |
c.842C>T |
p.Ala281Val |
Het |
Ex10 |
MS |
Pat |
rs63749950 |
1PR,3CRC |
|
P40/F |
|
MLH1 |
c.306+1G>A |
- |
Het |
Int3 |
SE |
Pat |
rs267607734 |
4CRC,1END,1Ov |
|
P36/F |
58/55 |
CHEK2 |
c.1427C>T |
p.Thr476Met |
Het |
Ex13 |
MS |
L.Pat |
rs142763740 |
2 BRE,1CRC |
|
P2/F |
38/37 |
MSH6 |
c.251C>T |
p.Ala84Val |
Het |
Ex10 |
MS |
VUS |
rs878853717 |
1LC, 1 LYM |
|
P4/M |
66/64 |
PMS2 |
c.2021C>T |
p.Thr671Met |
Het |
Ex12 |
MS |
VUS |
- |
1LRX,1LEU |
|
P8/F |
32/30 |
APC |
c.3625G>A |
p.Glu1209Lys |
Het |
Ex16 |
MS |
VUS |
rs201185479 |
1CRC |
|
P10/M |
48/47 |
APC |
c.4945A>G |
p.Ile1649Val |
Het |
Ex16 |
MS |
VUS |
rs772273122 |
1BLA,CRC,1CRC/BLA |
|
P14/F |
76/74 |
BRCA2 |
c.7985C>T |
p.Thr2662Met |
Het |
Ex18 |
MS |
VUS |
rs431825362 |
4CRC,1BRA |
|
SDHB |
c.178A>G |
p.Thr60Ala |
Het |
Ex2 |
MS |
VUS |
rs34599281 |
|||
|
P15/M |
75/70 |
ATM |
c.7522G>A |
p.Gly2508Arg |
Het |
Ex51 |
MS |
VUS |
rs754395517 |
- |
|
P16/F |
29/28 |
PALLD |
c.671_672delTGinsCA |
p.Met224Thr |
Het |
Ex2 |
MS |
VUS |
rs373066707 |
1LC,1PR |
|
P19/F |
39/38 |
APC |
c.3891T>G |
p.Asp1279Glu |
Het |
Ex16 |
MS |
VUS |
- |
1BRE |
|
P22/F |
53/51 |
MUTYH |
c.821G>A |
p.Arg274Gln |
Het |
Ex10 |
MS |
VUS |
rs149866955 |
2 PR,3CRC |
|
P22/F |
53/51 |
BRCA1 |
c.4342A>G |
p.Ser1448Gly |
Het |
Ex12 |
MS |
VUS |
rs80357486 |
2 PR,3CRC |
|
P23/F |
53/47 |
PALLD |
c.671_672delTGinsCA |
p.Met224Thr |
Het |
Ex2 |
MS |
VUS |
rs373066707 |
1CRC,1BRE,1LEU |
|
PMS1 |
c.1856+5G>T |
- |
Het |
Int9 |
SE |
VUS |
rs750408063 |
|||
|
ATR |
c.1732+4A>G |
- |
Het |
Int7 |
SE |
VUS |
- |
|||
|
P26/F |
61/60 |
ATM |
c.2932T>C |
p.Ser978Pro |
Het |
Ex20 |
MS |
VUS |
rs139552233 |
1END |
|
PMS2 |
c.2012C>T |
p.Thr671Met |
Het |
Ex12 |
MS |
VUS |
rs587780046 |
|||
|
P28/F |
37/35 |
MUTYH |
c.217G>A |
p.Glu73Lys |
Het |
Ex3 |
MS |
VUS |
rs1064794128 |
1LYM,1CRC |
|
P30/M |
46/45 |
POLD1 |
c.337G>A |
p.Gly113Arg |
Het |
Ex4 |
MS |
VUS |
rs1568618739 |
1CRC |
|
P31/F |
62/48 |
BRIP1 |
c.326A>G |
p.Asn109Ser |
Het |
Ex4 |
MS |
VUS |
rs587782734 |
3BRE,1END,1BRE/HL, 1KC,1ST,1Skin,1LRX,1 LEU |
|
PALLD |
c.671_672delTGinsCA |
p.Met224Thr |
Het |
Ex2 |
MS |
VUS |
rs373066707 |
|||
|
P33/M |
48/46 |
MSH6 |
c.14G>A |
p.Ser5Asn |
Het |
Ex1 |
MS |
VUS |
rs532585602 |
- |
|
P34/F |
56/55 |
SDHD |
c.283C>T |
p.Leu95= |
Het |
Ex3 |
SN |
VUS |
- |
4CRC |
|
P38/F |
35/33 |
BRCA2 |
c.1244A>G |
p.His415Arg |
Het |
Ex3 |
SN |
VUS |
rs80358417 |
2 LRX, 1 ST |
|
P, Patient; DX, Diagnose; AA, Aminoacid; Zyg, Zygosity; Het, Heterozygous; Hom, Homozygous; Loc, Localization; Ex, Exon; Int, Intron; Func, Function; FS, Frameshift; M, Missense; SE, Splice Effect; SN, Synonymous; Pat, Pathogenic; L.Pat, Likely Pathogenic; VUS, Variant of Uncertain Significance; dbSNP: a database of single nucleotide polymorphisms; CA, Cancer; PR, Prostate; LC, Lung; LRX, Larynx; CRC, Colorectal; End; Endometrium; HL, Hodgkin Lymphoma; ST, Stomach; BRE, Breast; BRA, Brain; PA, Pancreas; Ov,Ovarian; LEU, Leukemia; BLA, Bladder; LYM, Lymphoma; *Number of individuals with cancer in relatives; Transcript numbers of the causative genes identified in this study: NM_000179(MSH6), NM_001128425(MUTYH), NM_000249(MLH1), NM_000465(BARD1), NM_000051.3(ATM), NM_000535(PMS2), NM_007194.4(CHEK2), NM_000038(APC), NM_000059(BRCA2), NM_003000.3(SDHB), NM_001166108.1(PALLD), NM_007294(BRCA1), NM_000534.5(PMS1), NM_001184.4(ATR), NM_001256849.1(POLD1), NM_032043.2(BRIP1), NM_000179.2(MSH6), NM_003002(SDHD). |
||||||||||
In the three-generation pedigree analysis of all patients, it was seen that 6 patients were isolated cases, and relatives of the remaining patients were diagnosed with at least one cancer. Colon cancer was the most common malignancy in relatives diagnosed with cancer, and relatives of the patients had many types of heterogeneous cancer (Figure 2). There was no statistically significant difference between the mutation carriage status of the patients and the number of their relatives with cancer (p = 0.09).
DISCUSSION
Colorectal cancers are malignancies whose etiopathogenesis is contributed by biological, genetic, environmental, and behavioural factors; and the incidence is geographically and ethnically different all over the world. Age is an important risk factor in these cancers, and the majority of patients are diagnosed after age 50, and the incidence increases dramatically with each decade after age 50.7 In recent years, there has been an increase in early age colon cancers in the West and many Asian and African countries. For example, it is thought that nearly half of the CRC patients in Pakistan are diagnosed at a young age.8 In a study from the same country, it was found that hereditary non-polyposis colorectal carcinoma was seen at an earlier age in those whose parents were relatives themselves, and had relative-history of cancer.9 The early age of onset in CRCs suggests that there may be an inherited predisposition. Genetic causes are thought to play a role in the etiopathogenesis of one out of every five CRC cases diagnosed under the age of 50.10
In this study, the mean age at diagnosis of colorectal cancer was 48.7 ± 12.85 years. With statistical analysis, no significant difference was detected between those who have mutation and those who do not, in terms of age at diagnosis of cancer. A causally inherited (germline) cancer susceptibility gene was detected in 24.4% of patients with CRC and the genetic background of their diseases was clarified. Pathogenic/possibly pathogenic gene variants in these 10 patients were located on the dominantly autosomal inherited MLH1, MSH6, CHEK2, PMS2, and MUTYH genes.
Approximately 30% of CRC cases, which are among the common malignancies, have been shown in the literature through studies of relatives and twins.11,12 Approximately, 5% of these cases are associated with well-characterised, highly penetrating hereditary mutations. The etiology of the remaining 20-30% of CRCs, other than these well-characterised syndromes, is not fully understood; and it is thought that they may result from single-gene changes with low penetration and more commonly detected.13 A thorough understanding of the genetics of hereditary CRCs is important to identify individuals at risk, improve cancer surveillance and prevention strategies, and develop better diagnostic and treatment approaches.
In this study, 59 germline cancer susceptibility genes of 41 Turkish patients diagnosed with CRC were examined. Eight (80%) of 10 patients with mutations were diagnosed with HNPCC because they had germline MMR causal gene variants. Patients who were diagnosed with HNPCC as a result of genetic analysis were given genetic counselling on cancer risks associated with this syndrome.
HNPCC (also known as lynch syndrome), the most common cause of inherited CRC, accounts for about 2-4% of all CRCs. In this syndrome, there is also an increased risk of non-colon cancers such as cancers of endometrium, ovaries, stomach, small intestine, hepatobiliary system, urinary tract and central nervous system. The lifetime risk of CRC in these patients is estimated to be 50-80% and the risk of developing endometrial cancer varies between 40-60%.4,14,15 The overall prevalence of the disease is thought to be 1/440. The main function of the MLH1, MSH2, MSH6, PMS2 and EPCAM genes (also known as MMR genes), that cause this syndrome, is to help maintain DNA's effectiveness by correcting DNA replication errors. Of these genes, mutations in MLH1 and MSH2 make up about 90% of HNPCC cases. Mutations in MSH6 cause about 10% of cases. PMS2 mutations, on the other hand, are extremely rare.16 It has been reported that MLH1 and MSH2 mutations carry a higher risk of cancer development compared to MSH6 and PMS2. Although not belonging to the MMR gene family, germline deletions seen in the EPCAM gene can lead to inactivation of the MSH2 gene, and this condition may be encountered in approximately 1-3% of HNPCC cases. Recent data in the literature suggest that EPCAM deletions may primarily cause CRC.17,18
In studies conducted with CRC patients from different populations in the literature, HNPCC-related genes were investigated, and causal variants were found in various ratios in these cases. The mutation carriage rate in HNPCC-related genes in patients with CRC from countries such as the USA, Pakistan, Korea, China, Singapore, Poland, and Brazil varied between 28-38%.19 In a study conducted in Taiwan, this rate was found to be around 60%.20
As a result of the genetic analysis performed, a causal variant in the CHEK2 gene of a patient was detected in heterozygous condition. The CHEK2 gene is involved in the formation of the CHEK2 protein, a protein kinase that is activated in response to DNA damage. This protein is involved in DNA repair, cell cycle arrest, control, and apoptosis. The CHEK2 gene has been associated with hereditary breast and CRC predisposition in the OMIM database. In addition, heterozygous mutations of this gene are responsible for Li-Fraumeni-like syndrome (LFS2), a highly penetrating familial cancer syndrome. In the individual and/or family history, especially breast, colon, prostate, or other, cancers may be associated with CHEK2 pathogenic variants.21 In the patient in this study, the detected CHEK2 gene c.1427C>T variant replaces threonine with methionine at 476 codons of the protein of interest (p.Thr476Met). This variant [c.1427C>T (p.Thr476Met)] has been reported in population databases and, in many patients with breast, endometrium, ovary, colorectal, and prostate cancers in the literature.22,23
One of the patients in the study was found to be homozygous for the c.884 C>T causative variant in the MUTYH gene. This variant is found in population databases at a fairly low frequency (rs374950566, ExAC <0.01%). It has been reported that, this variant is observed in both homozygous and combined heterozygous conditions in individuals diagnosed with MAP in the literature.24 MAP is an autosomal recessively inherited syndrome caused by a germline mutation of both alleles of the MUTYH gene (also known as the MYH gene). While most patients have 10 to several hundred adenomas, some patients have early onset CRC and no polyp is found. Colorectal polyps usually develop around the age of 40. The risk of developing CRC in MAP patients is 19% by age 50 and 43% by age 60. The mean age of onset of CRC in these individuals is 48.25 It has been reported that there is an increased risk in relatives of MAP patients with heterozygous MUTYH mutations when compared with their relatives of sporadic CRC. Gastric and duodenal polyps are seen in 11-17% of these patients. The lifetime risk of duodenal cancer is approximately 4% in MAP patients, compared to the general population. Although the risk of many extraintestinal malignancies has also been reported to double, including ovarian, bladder, skin, and possibly breast cancer; for these extraintestinal malignancies, intensive surveillance measures are not recommended.24
The sample size of this study was not large enough, segregation analysis could not be performed for all cancer relatives in pedigree analysis in terms of causal gene variants, and environmental factors, that could cause cancer susceptibility, could not be ruled out. In addition, although a large number of genes of the patients have been analysed, all candidate genes that may produce a predisposition specific to our population, have not been examined.
CONCLUSION
The presence of possible genetic predisposition factors in the etiopathogenesis and pathophysiology of CRC patients should be investigated. NGS-based screening has high clinical sensitivity, and its use in the detection of mutation in CRC will aid in understanding its genetic causes. Identifying that these patients carry a causal variant in their cancer-related genes and diagnosing a hereditary cancer syndrome will provide information on other primary cancer risks that may develop; and direct them to a close follow-up strategy. This awareness will make it possible for the sick individual to be included in an intensive cancer screening, monitoring and prevention programme.
ETHICAL APPROVAL:
This observational cohort study was approved by local Ethics Committee.
PATIENTS’ CONSENT:
Since this study was designed retrospectively, the data presented were collected from the hospital archives after the approval of the Ethics Committee.
CONFLICT OF INTEREST:
The authors declared no conflict of interest.
AUTHORS’ CONTRIBUTION:
All authors have done conception or design of the work or the acquisition and analysis, or interpretation of data for the work, drafting the work or revising it critically for important intellectual content, and gave final approval of the version to be published.
REFERENCES
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca J Clin 2018; 68(6):394-424. doi: 10.3322/ caac.21492
- Ferlay J, Ervik M, Lam F, Colombet M, Mery L, Piñeros M, et al. Global cancer observatory: Cancer today. Lyon, France: International Agency for Research on Cancer. 2018;
- Liu Q, Tan YQ. Advances in identification of susceptibility gene defects of hereditary colorectal cancer. J Cancer 2019; 10(3):643. doi: 10.7150/jca.28542.
- Cetinkaya K, Yuce E. Lynch syndrome in patients treated for endometrial cancer. Euro J Gynaecol Oncol 2017; 38(4): 607-13.
- Clark S. Management of genetically determined colorectal cancer. Surgeon 2019; 17(3):165-71. doi: 10.1016/j.surge. 2019.03.003.
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med 2015; 17(5):405-23. doi: 10.1038/gim.2015.30.
- Tunio MA, Rafi M, Hashmi A. Hereditary nonpolyposis colorectal cancer in Pakistan: Results of a pilot study. Pak J Med Sci 2011; 27(2):339-43.
- Bhurgri Y, Khan T, Kayani N, Ahmad R, Usman A, Bhurgri A, et al. Incidence and current trends of colorectal malignancies in an unscreened, low risk population. Asian Pacific J Cancer Prevention 2011; 12(3):703-8.
- Javaid N. Frequency of hereditary nonpolyposis colorectal cancers in Punjab Pakistan. Annals Oncol 2017; 28:iii91.
- Muller C, Ihionkhan E, Stoffel EM, Kupfer SS. Disparities in early-onset colorectal cancer. Cells 2021; 10(5):1018. doi: 10.3390/cells10051018.
- Hampel H. Genetic testing for hereditary colorectal cancer. Surg Oncol Clin 2009; 18(4):687-703.
- Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, et al. Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland. N Eng J Med 2000; 343(2):78-85. doi: 10.1056/NEJM200007133430201.
- Jasperson KW, Tuohy TM, Neklason DW, Burt RW. Hereditary and familial colon cancer. Gastroenterol 2010; 138(6):2044-58. doi: 10.1053/j.gastro.2010.01.054.
- Tanriverdi O, Yilmaz EM, Menekse S, Cokmert S, Oktay E, Pilanci KN, et al. The recurrence with isolated intra-abdominal lymph node in patients with colorectal cancer: A study of the Turkish descriptive oncological researches group (intra-abdominal lymph node and colon cancer). J Oncol Sci 2016; 2(2-3):48-52. doi.org/10.1016/j.jons. 2016.07.001
- Win AK, Lindor NM, Young JP, Macrae FA, Young GP, Williamson E, et al. Risks of primary extracolonic cancers following colorectal cancer in lynch syndrome. J Natl Cancer Ins 2012; 104(18):1363-72. doi: 10.1093/jnci/djs351.
- Ma H, Brosens LA, Offerhaus GJA, Giardiello FM, de Leng WW, Montgomery EA. Pathology and genetics of hereditary colorectal cancer. Pathol 2018; 50(1):49-59. doi: 10.1016/j.pathol.2017.09.004.
- Weissman SM, Burt R, Church J, Erdman S, Hampel H, Holter S, et al. Identification of individuals at risk for Lynch syndrome using targeted evaluations and genetic testing: National Society of Genetic Counselors and the Collaborative Group of the Americas on Inherited Colorectal Cancer joint practice guideline. J Genet Couns 2012; 21(4):484-93. doi: 10.1007/s10897-011-9465-7.
- Giardiello FM, Allen JI, Axilbund JE, Boland CR, Burke CA, Burt RW, et al. Guidelines on genetic evaluation and management of Lynch syndrome: A consensus statement by the US Multi-society task force on colorectal cancer. Dis Colon Rectum 2014; 57(8):1025-48. doi: 10.1097/DCR. 000000000000000.
- Rashid MU, Naeemi H, Muhammad N, Loya A, Lubiński J, Jakubowska A, et al. Prevalence and spectrum of MLH1, MSH2, and MSH6 pathogenic germline variants in Pakistani colorectal cancer patients. Hereditary Cancer Clin Prac 2019; 17(1):1-16. doi: 10.1186/s13053-019-0128-2.
- Kamiza AB, Hsieh LL, Tang R, Chien HT, Lai CH, Chiu LL, et al. Risk factors associated with colorectal cancer in a subset of patients with mutations in MLH1 and MSH2 in Taiwan fulfilling the Amsterdam II criteria for Lynch syndrome. PloS One 2015; 10(6):e0130018. doi: 10.1371/journal.pone. 0130018.
- Schneider K, Zelley K, Nichols KE, Garber J. Li-fraumeni syndrome. 2019.
- DeRycke MS, Gunawardena S, Balcom JR, Pickart AM, Waltman LA, French AJ, et al. Targeted sequencing of 36 known or putative colorectal cancer susceptibility genes. Mol Genet Genomic Med 2017; 5(5):553-69. doi: 10.1002/mgg3.317.
- Pritzlaff M, Summerour P, McFarland R, Li S, Reineke P, Dolinsky JS, et al. Male breast cancer in a multi-gene panel testing cohort: Insights and unexpected results. Breast Cancer Res Treat 2017; 161(3):575-86. doi: 10.1007/ s10549-016-4085-4.
- Vogt S, Jones N, Christian D, Engel C, Nielsen M, Kaufmann A, et al. Expanded extracolonic tumor spectrum in MUTYH-associated polyposis. Gastroenterol 2009; 137(6): 1976-85.e10. doi: 10.1053/j.gastro.2009.08.052.
- Nielsen M, Morreau H, Vasen HF, Hes FJ. MUTYH-associated polyposis (MAP). Crit Rev Oncol Hematol 2011; 79(1):1-16. doi: 10.1016/j.critrevonc.2010.05.011.