A Case of Foveoschisis Associated with Ornithine Aminotransferase Deficiency and Gyrate Atrophy
By Sehnaz Ozcaliskan1, Sevcan Balci2, Ozgur Artunay1Affiliations
doi: 10.29271/jcpsp.2021.11.1354ABSTRACT
Gyrate atrophy is a metabolic disorder characterised by typical progressive circular chorioretinal atrophy, myopia and early developmental cataract. The disease is caused by deficiency of ornithine aminotransferase (OAT) enzyme. Although OAT is expressed in most tissues of the body, but the main target of the disease appears to be the retina. A case is presented here of a 21-year woman, who came to our clinic with the complaint of decline in central vision for eight months. She had progressive poor night vision and was diagnosed with OAT deficiency five years ago. Her systemic history was unremarkable, except for femoral deep vein thrombosis (DVT) which occurred two years ago. Laboratory tests performed at that time had revealed elevated serum ornithine and low serum lysin levels. Optic coherence tomography (OCT) scans showed foveoschisis bilaterally. In summary, gyrate atrophy may present as macular involvement in the form of foveoschisis and may lead to impaired central vision.
Key Words: Foveoschisis, Gyrate atrophy, Ornithine aminotransferase.
INTRODUCTION
Ornithine aminotransferase (OAT) deficiency is a rare, autosomal recessive congenital metabolic disorder characterised by choroidal and gyrate atrophy (GA) of the retina, and elevated serum ornithine levels. Patients with GA commonly present with a decline in visual acuity and nictalopia. Typical circular chorioretinal atrophic areas surround the entire retina with sharp margins and hyperpigmented edges. These findings may also be accompanied by axial myopia and posterior subcapsular cataract.1 To reduce serum ornithine levels in patients with hyperornithinemia, arginine-restricted low-protein diet and vitamin B6 are used. In late stages of the disease, macular involvement may be seen.2 Hereby, we report a case of GA accompanied by foveoschisis and discuss the plausible mechanisms that lead to foveoschisis.
CASE REPORT
A 21-year old woman presented to our clinic with the complaint of decline in central vision for eight months. She had progressive poor night vision and was diagnosed with OAT deficiency five years ago. She did not have genetic testing and her family history was unremarkable. She also had a history of femoral deep vein thrombosis (DVT) two years ago. Her laboratory tests performed at that time revealed normal results, except for elevated serum ornithine (489.80 µmol/L) and low serum lysin levels (93.60 µmol/L). The remaining of the systemic and neurologic examinations were unremarkable.
On her ophthalmologic evaluation, best corrected visual acuity was 20/50 with -3.75 diopters (D) in the right eye and 20/63 with -4.50 D in the left eye. Intraocular pressures were in normal range bilaterally. The axial length (AL) measurement was 24.78 mm in the right eye and 24.86 mm in the left eye. Biomicroscopic examination revealed no signs other than bilateral posterior subcapsular cataracts, more prominent in her left eye. Fundus examination showed sharp typical chorioretinal atrophic areas extending from the posterior pole to the periphery (Figure 1). Optical coherence tomography (OCT) imaging revealed increased macular thickness and multiple hyporeflective spaces surrounded by septae in the inner nuclear layer which were consistent with foveoschisis bilaterally (Figure 2). Fundus fluorescein angiography (FFA) showed no leakage in early and late phases. In fundus autofluorescence (FAF) imaging, hypoautofluorescence areas compatible with the sharp atrophy areas in midperiphery and central demarcated hyperautofluorescence perimeter, were noted (Figure 3). Her full field electroretinography (ERG) results were unremarkable. Based on her history and ophthalmological examination showing the typical chorioretinal atrophic areas, she was clinically diagnosed with GA. She was advised 300 mg pyridoxine supplementation daily to lower plasma ornithine levels, but she refused to take any medication due to her gastrointestinal disturbances.
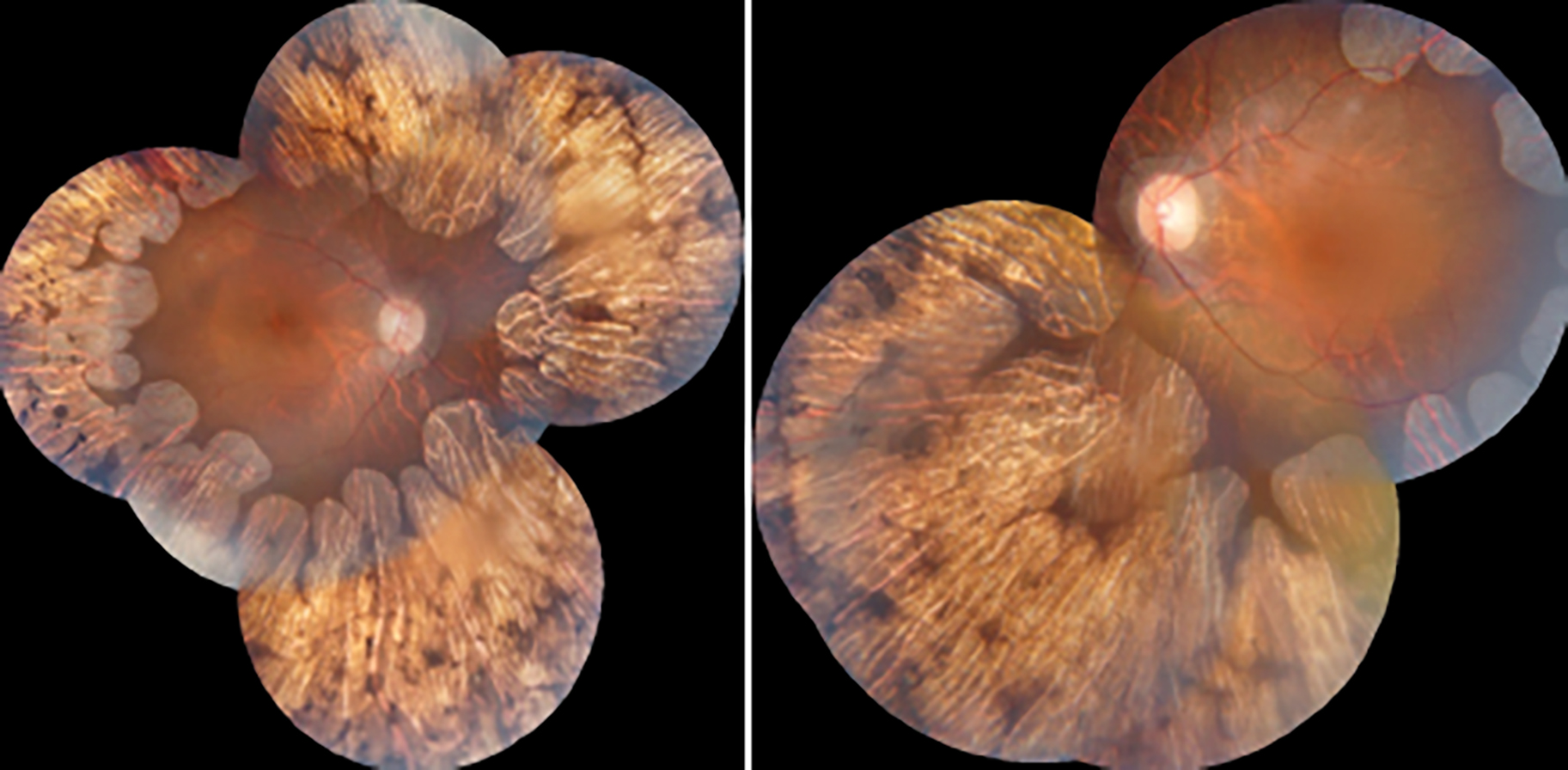 Figure 1: Color fundus photographs showing sharp chorioretinal atrophic areas extending from the posterior pole to the periphery.
Figure 1: Color fundus photographs showing sharp chorioretinal atrophic areas extending from the posterior pole to the periphery.
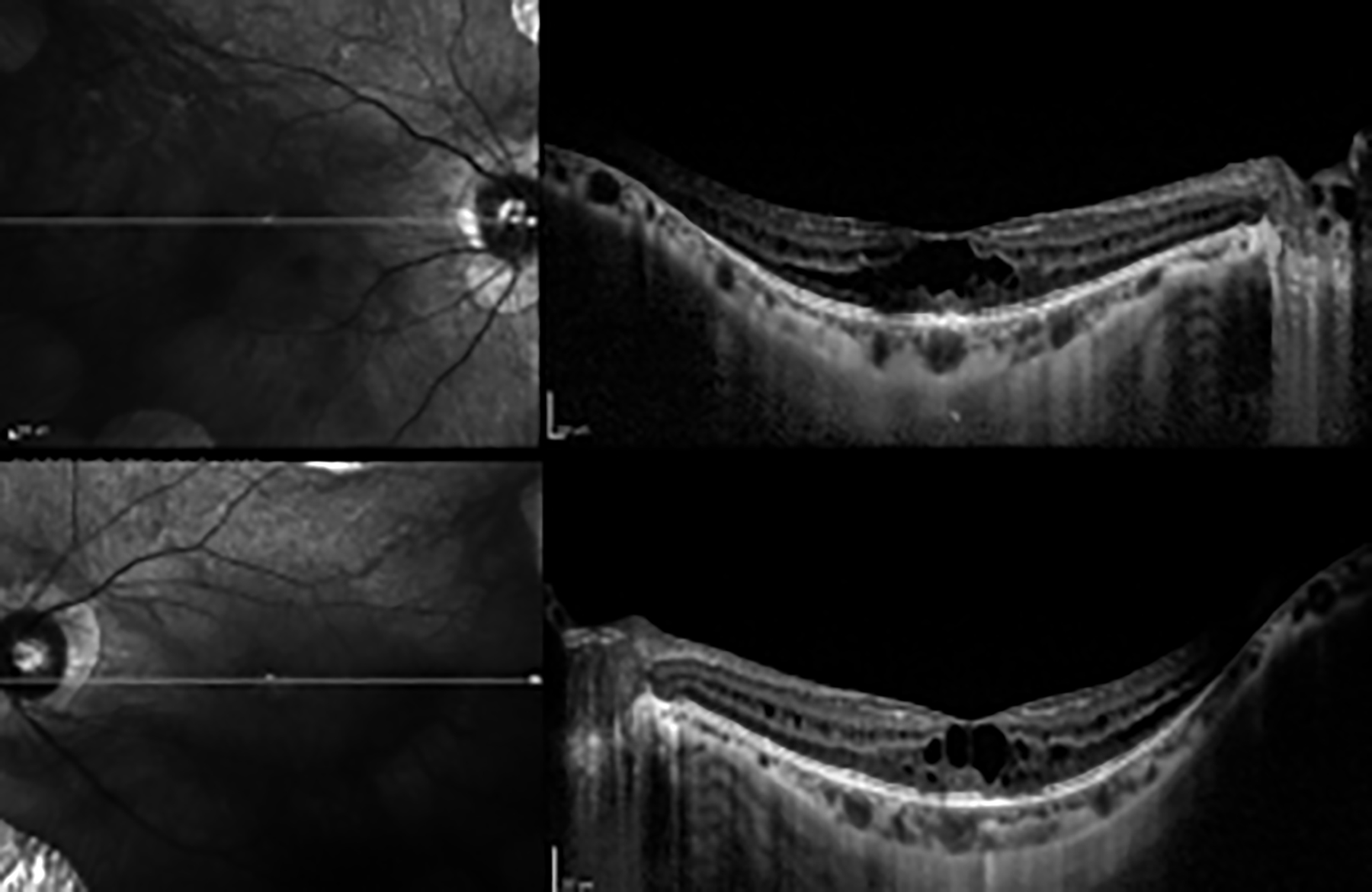 Figure 2: Optical coherence tomography (OCT) imaging revealing increased macular thickness and multiple hyporeflective spaces surrounded by septae in the inner nuclear layer.
Figure 2: Optical coherence tomography (OCT) imaging revealing increased macular thickness and multiple hyporeflective spaces surrounded by septae in the inner nuclear layer.
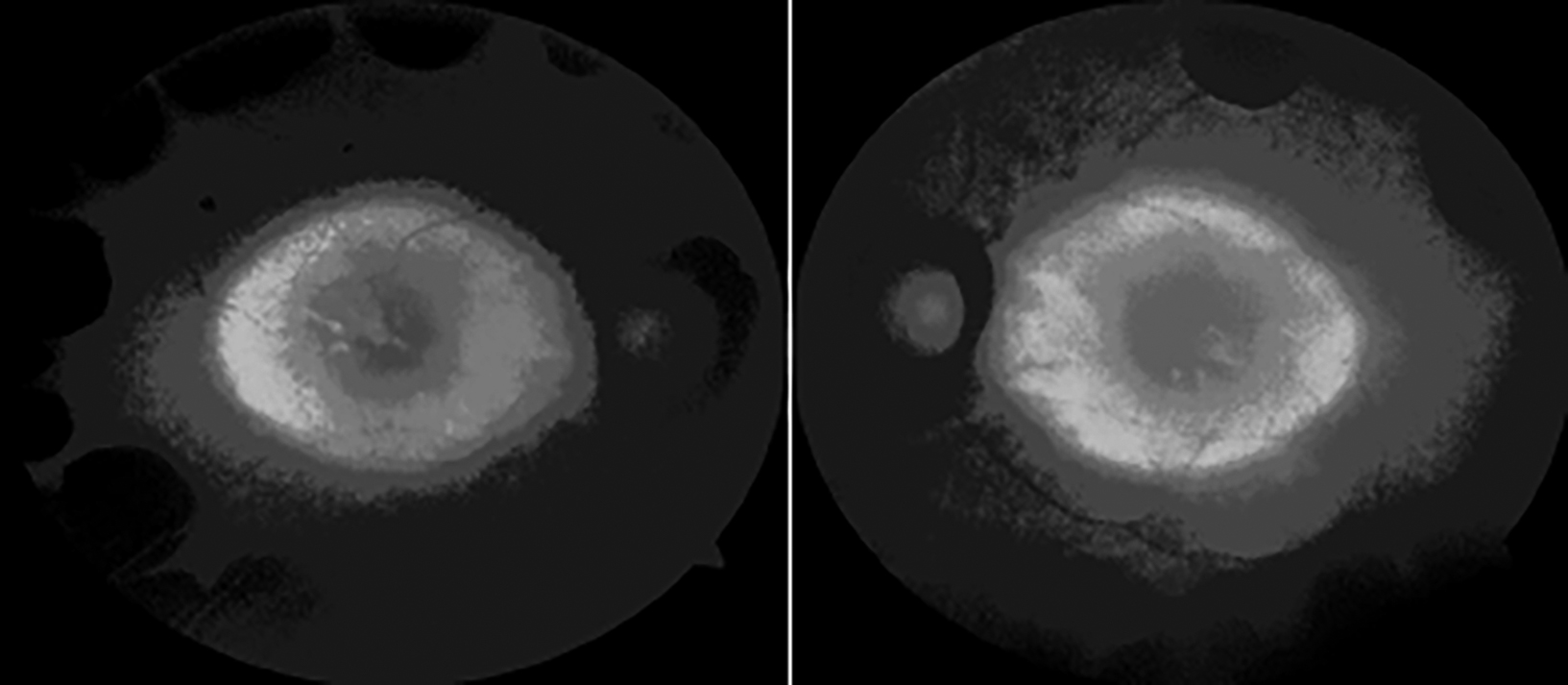 Figure 3: Fundus autofluorescence showing hypoautofluorescence areas compatible with the sharp atrophy areas in midperiphery and central demarcated hyperautofluorescence perimeter.
Figure 3: Fundus autofluorescence showing hypoautofluorescence areas compatible with the sharp atrophy areas in midperiphery and central demarcated hyperautofluorescence perimeter.
DISCUSSION
GA is an autosomal recessive metabolic disorder with hyperornithinemia due to the OAT enzyme deficiency. In this rare condition, retinal and choroidal atrophy develops with unknown pathogenesis. Because the typical ophthalmoscopic appearance of atrophic areas resembles brain gyri, the disease is called as GA. The initial symptom, the night blindness, is related to multiple sharp areas of chorioretinal atrophy in the midperipheral retina. At later stages, atrophic areas merge and progress to the posterior pole. These typical areas of atrophy are accompanied by other ophthalmologic findings, such as myopia, cataract and vitreous opacities.3 Macular involvement in this condition has been reported as macular edema, macular hole, choroidal neovascularization and foveoschisis.4
In GA, foveoschisis may cause retinal defects and irregularity of the retinal layers, resulting in a sudden decrease in visual acuity.5 OAT enzyme is expressed in most tissues and ornithine is the key reactant in amino-acid catabolism and urea cycle. In the presence of OAT deficiency, retinal pigment epithelial pump function impairment and Müller cell dysfunction may develop.6 Consequently, extracellular fluid accumulation and foveoschisis may occur in the neural retina. Another factor to be considered in the development of foveoschisis is the presence of myopia accompanying the disease. Myopic foveoschisis is a finding in approximately 10-14.6% cases of pathologic myopia.7,8 Many factors such as abnormal vitreoretinal interface and posterior vitreous detachment, progressive scleral ectasia and staphyloma formation are thought to play a role in the development of myopic foveoschisis.9 However, our patient had moderate myopia with AL values within normal range, bilaterally.
Arginine, is a semi-essential amino-acid and precursor of ornithine. It plays a role in metabolic pathways, particularly in urea cycle and nitric oxide synthesis. Nitric oxide acts as a potent vasodilator and an inhibitor in coagulation cascade, while urea cycle eliminates toxic ammonia from the body. Our patient had a history of DVT, which suggests a plausible alteration in these metabolic pathways. But, no specific predisposing factor for thrombosis was detected in the tests performed at that time. It is likely that she developed DVT coincidentally. Previous reports described systemic features of OAT deficiency and GA, such as thin and rare hairs, mental retardation with diffuse brain atrophy, electro‐encephalographic abnormalities and muscular weakness.10 Dietary protein and arginine restriction may reduce blood ornithine levels in GA. Chronic reduction of ornithine with arginine-restricted diet, initiated at an early age, has been reported to slow the progression of chorioretinal dystrophy.11 Vitamin B6 derivative, pyridoxine, is a cofactor of the OAT enzyme and may lower elevated plasma ornithine levels when high doses are administered.12 However, the efficacy of these supplementary treatments in patients with GA is unclear.
In conclusion, GA may present as macular involvement in the form of foveoschisis and may lead to impaired central vision. Although OAT deficiency frequently presents with eye involvement, ophthalmologists should be aware that the disease is a systemic disorder.
PATIENT’S CONSENT:
Informed consent was obtained from the patient to publish the data concerning this case.
CONFLICT OF INTEREST:
The authors declared no conflict of interest.
AUTHORS’ CONTRIBUTION:
SO, SB, OA: Conception or design of the work, or the acquisition, analyses, or interpretation of data for the work.
REFERENCES
- Caruso RC, Nussenblatt RB, Csaky KG, Valle D, Kaiser-Kupfer MI. Assessment of visual function in patients with gyrate atrophy who are considered candidates for gene replacement. Arch Ophthalmol 2001; 119(5):667-9. doi: 10.1001/archopht.119.5.667.
- Tripathy K, Chawla R, Sharma YR, Gogia V. Ultrawide field fluorescein angiogram in a family with gyrate atrophy and foveoschisis. Oman J Ophthalmol 2016; 9(2):104-6. doi: 10.4103/0974-620X.184529.
- Büyüktortop N, Alp MN, Sivri S, Coşkun T, Kural G. Gyrate atrophy of the choroid and retina: A case report. Turk J Pediatr 2011; 53(1):94-6.
- Shah VS. Gyrate Atrophy. In: Medina C, Townsend J, Singh A. (eds) Manual of retinal diseases. 2016; Springer, Cham.
- Tekin K, Ozates S, Teke MY. A rare association of foveoschisis with gyrate atrohy. DJO 2017; 28(1):58-9.
- Wang T, Milam AH, Steel G, Valle D. A mouse model of gyrate atrophy of the choroid and retina. Early retinal pigment epithelium damage and progressive retinal degeneration. J Clin Invest 1996; 97(12):2753-62. doi: 10.1172/JCI118730.
- Silva R. Myopic maculopathy: A review. Ophthalmologica 2012; 228(4):197-213. doi: 10.1159/000339893.
- Henaine-Berra A, Zand-Hadas IM, Fromow-Guerra J, García-Aguirre G. Prevalence of macular anatomic abnormalities in high myopia. Ophthalmic Surg Lasers Imaging Retina 2013; 44(2):140-4. doi: 10.3928/23258160-20130219-01
- Gohil R, Sivaprasad S, Han LT, Mathew R, Kiousis G, Yang Y. Myopic foveoschisis: A clinical review. Eye (Lond) 2015; 29(5):593-601. doi: 10.1038/eye.2014.311.
- Kaiser-Kupfer MI, Kuwabara T, Askanas V, Brody L, Takki K, Dvoretzky I, et al. Systemic manifestations of gyrate atrophy of the choroid and retina. Ophthalmology 1981; 88(4):302-6. doi: 10.1016/s0161-6420(81)35030-1.
- Michaud J, Thompson GN, Brody LC, Steel G, Obie C, Fontaine G, et al. Pyridoxine-responsive gyrate atrophy of the choroid and retina: Clinical and biochemical correlates of the mutation A226V. Am J Hum Genet 1995; 56(3): 616-22.
- Kaiser Kupfer MI, Caruso RC, Valle D. Gyrate atrophy of the choroid and retina. Long-term reduction of ornithine slows retinal degeneration. Arch Ophthalmol 1991; 109(11): 1539-48.